-
RNA-seq——四、根据序列比对结果筛选差异基因
写在前面——经过前面的一系列分析,我们得到了几个counts数据,接下来就需要根据这些数据来进行分析。本文使用Rstudio,从序列比对结果中筛选出差异基因,目的是(根据不同基因的表达量)找出实验组与对照组的差异。本文使用的数据见RNA-seq——上游分析练习(数据下载+hisat2+samtools+htseq-count)
参考:
RNA-seq(6): reads计数,合并矩阵并进行注释
RNA-seq(7): DEseq2筛选差异表达基因并注释(bioMart)1. 合并矩阵并进行注释
rm(list = ls()) options(stringsAsFactors = FALSE) # 读取数据 control_1 <- read.table("SRR3589959.count", col.names = c("gene_id", "control_1")) control_2 <- read.table("SRR3589961.count", col.names = c("gene_id", "control_2")) treat_1 <- read.table("SRR3589960.count", col.names = c("gene_id", "treat_1")) treat_2 <- read.table("SRR3589962.count", col.names = c("gene_id", "treat_2")) # 将数据合并 raw_count <- merge(merge(control_1, control_2, by = "gene_id"), merge(treat_1, treat_2, by = "gene_id")) # head(raw_count) # 去除无用信息 raw_count_filt <- raw_count[-1:-5,] # 修改行名 ENSEMBL <- gsub("\\.\\d*", "", raw_count_filt$gene_id) row.names(raw_count_filt) <- ENSEMBL- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
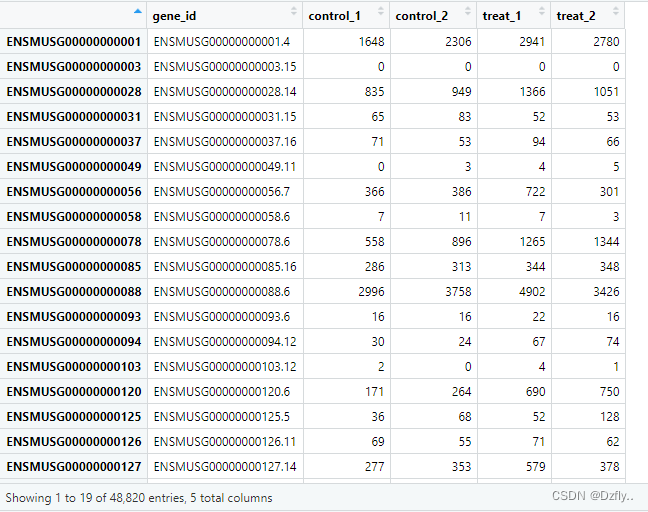
处理完的raw_count_filt:
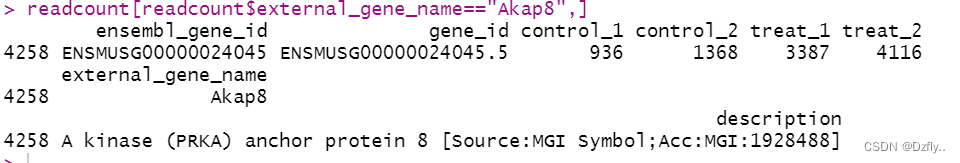
# 将ensembl_gene_id转化为gene_symbol library(biomaRt) library(curl) my_ensembl_gene_id <- row.names(raw_count_filt) mart <- useDataset("mmusculus_gene_ensembl", useMart("ensembl")) mms_symbols <- getBM(attributes = c("ensembl_gene_id", "external_gene_name", "description"), filters = "ensembl_gene_id", values = my_ensembl_gene_id, mart = mart) # 方便按照ensembl_gene_id来合并两个数据集 raw_count_filt <- cbind(ENSEMBL, raw_count_filt) colnames(raw_count_filt)[1] <- c("ensembl_gene_id") # 合并 readcount <- merge(raw_count_filt, mms_symbols, by = "ensembl_gene_id") # 保存 write.csv(readcount, file = "readcount.csv") # 查看Akap8的表达情况 readcount[readcount$external_gene_name=="Akap8",] # 整理数据,方便后续使用 rownames(readcount) <- readcount$ensembl_gene_id mycounts <- readcount[,3:6] write.csv(mycounts, file = "mycounts.csv")- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29

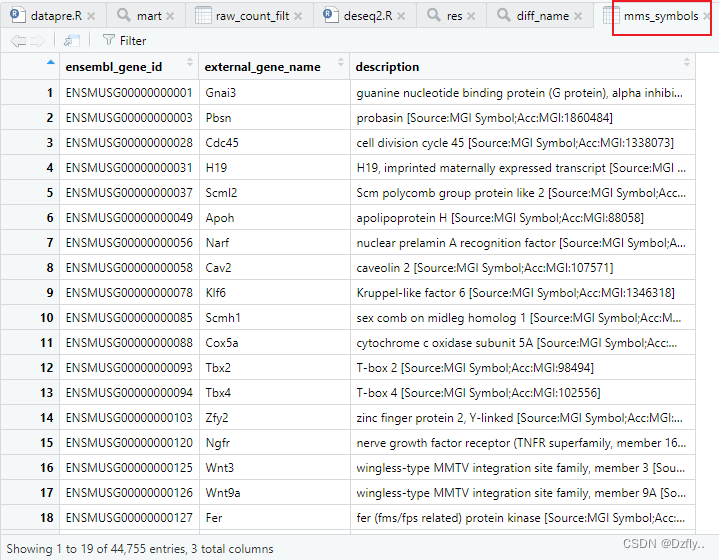
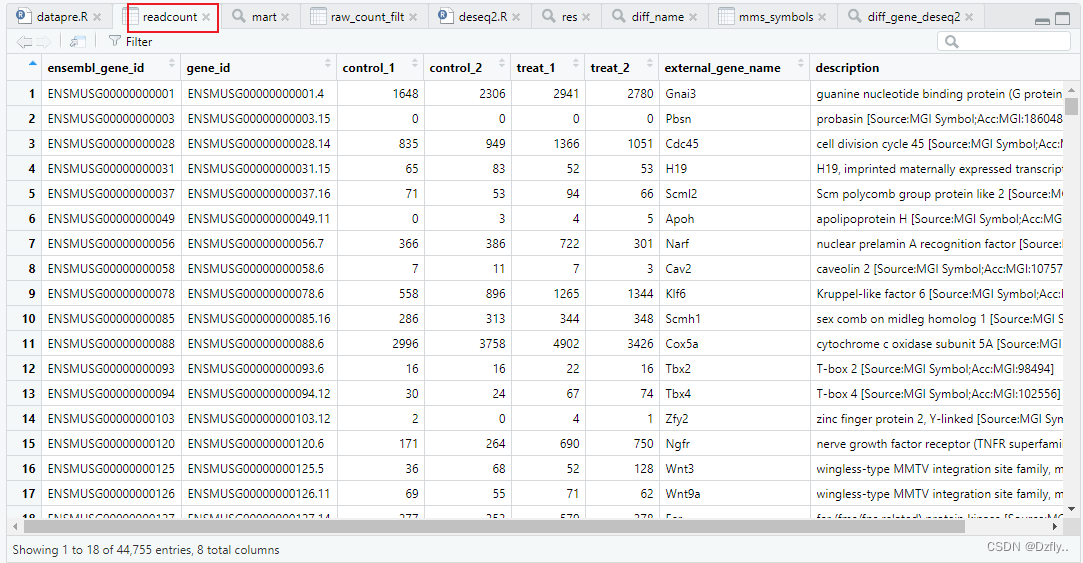

整理好的数据包含Ensembl ID以及每组的基因比对结果。2. 筛选差异基因(DESeq2)
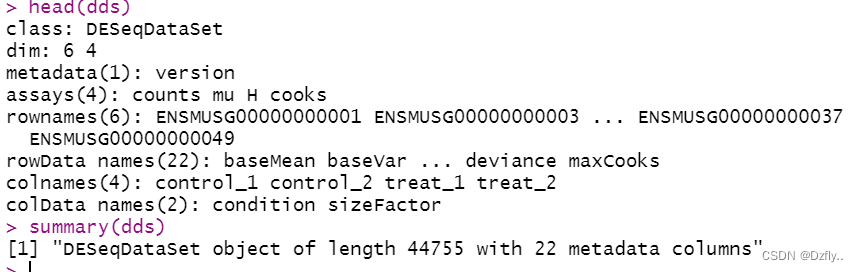
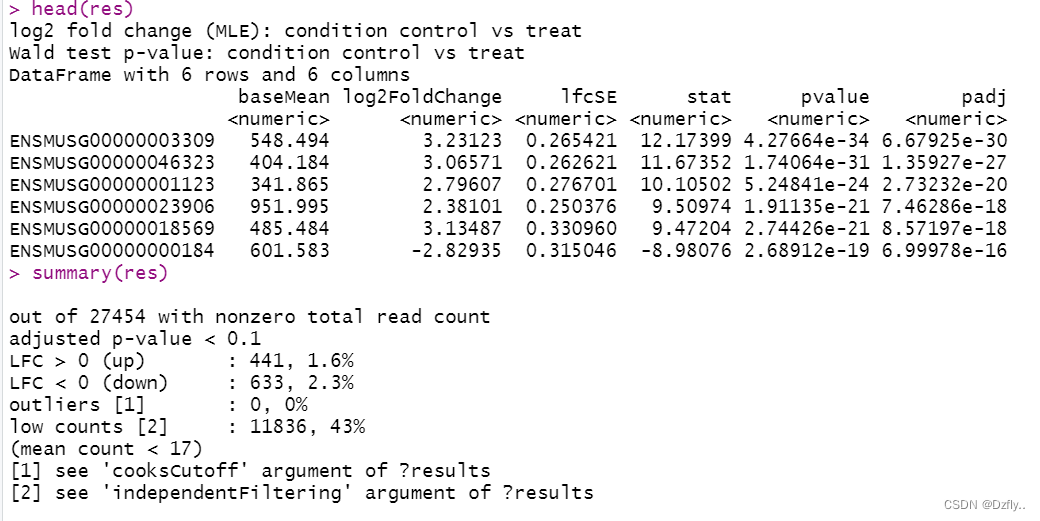
rm(list = ls()) library(tidyverse) library(DESeq2) # 读取上一步整理好的数据 mycounts <- read.csv("mycounts.csv") rownames(mycounts) <- mycounts$X mycounts <- mycounts[,-1] # 设置factor condition <- factor(c(rep("control", 2), rep("treat", 2)), levels = c("control", "treat")) # 生成colData colData <- data.frame(row.names = colnames(mycounts), condition) # 使用DESeq2分析 dds <- DESeqDataSetFromMatrix(mycounts, colData, design = ~ condition) dds <- DESeq(dds) res <- results(dds, contrast = c("condition", "control", "treat")) res <- res[order(res$pvalue),] # summary(res) # table(res$padj<0.05) write.csv(res, file = "all_results.csv") # 筛选差异基因 # P和log2FC的值可以自己设置,值不同,筛选出来的差异基因数目也不同 diff_gene_deseq2 <- subset(res, padj < 0.05 & abs(log2FoldChange) >1) write.csv(diff_gene_deseq2, file = "DEG_treat_vs_control.csv") # 将Ensembl ID转化为Gene Symbol # 方法一 # library(clusterProfiler) # library(org.Mm.eg.db) # # name <- bitr(rownames(diff_gene_deseq2), fromType = "ENSEMBL", toType = "SYMBOL", # OrgDb = "org.Mm.eg.db") # 方法二 library(biomaRt) library(curl) my_ensembl_gene_id <- row.names(diff_gene_deseq2) mart <- useDataset("mmusculus_gene_ensembl", useMart("ensembl")) mms_symbols <- getBM(attributes = c("ensembl_gene_id", "external_gene_name", "description"), filters = "ensembl_gene_id", values = my_ensembl_gene_id, mart = mart) ensembl_gene_id <- rownames(diff_gene_deseq2) diff_gene_deseq2 <- cbind(ensembl_gene_id, diff_gene_deseq2) colnames(diff_gene_deseq2)[1] <- c("ensembl_gene_id") # 得到最终结果 diff_name <- merge(diff_gene_deseq2, mms_symbols, by = "ensembl_gene_id") diff_name[diff_name$external_gene_name=="Akap8",]- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59

至此,我们便得到了差异基因,之后就可以根据这些差异基因,进行基因的富集分析,可以更加细致的了解到实验组的变化。更重要的是,之后可以作出一些精美的图片,帮助我们发文章~~/(ㄒoㄒ)/~~ -
相关阅读:
Mybatis的特性详解——动态SQL
Selenium自动化测试之Selenium IDE
vue ant 隐藏列
【uniapp微信小程序+springBoot(binarywang)
Java 基础高频面试题(2022年最新版)
扩展卡尔曼滤波EKF
【计算机网络】应用层自定义协议
C 语言 时间函数使用技巧(汇总)
计算机设计大赛 深度学习驾驶行为状态检测系统(疲劳 抽烟 喝水 玩手机) - opencv python
开发3年入职饿了么P6,全靠这份MyBatis学习笔记了
- 原文地址:https://blog.csdn.net/narutodzx/article/details/126807910
