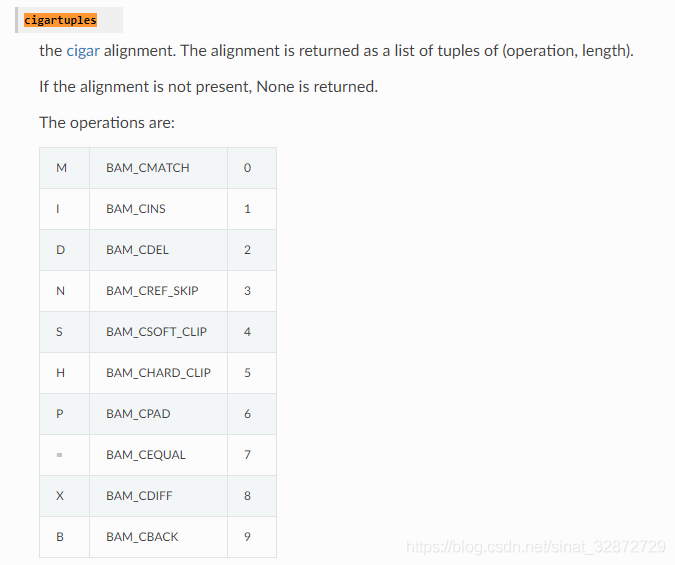
-
【bioinfo】sam文件可选区域字段(Optional Feild)含义
写在前面
上面两个链接是关于sam文件格式的官方说明文档,第一个包含所有字段,但对可选字段的说明比较简略,第二个是对可选字段的详细说明。这里根据官方文档,说明一下可选字段的具体含义。
下面是sam文件的示例,其中第12列及以之后(红色框)就是可选字段:
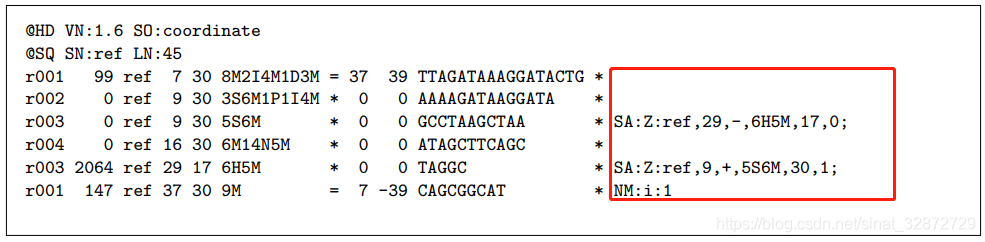
可选字段说明
可选字段的构成格式: TAG:TYPE:VALUE,例如,上面sam文件示例,可选字段
SA:Z:ref,9,+,5S6M,30,1;中,SA对应TAG,Z对应TYPE,ref,9,+,5S6M,30,1对应VALUE。其中,TYPE字符对应5中类型A (character), B (general
array), f (real number), H (hexadecimal array), i (integer), or Z (string)。其对应的VALUE正则匹配如下:

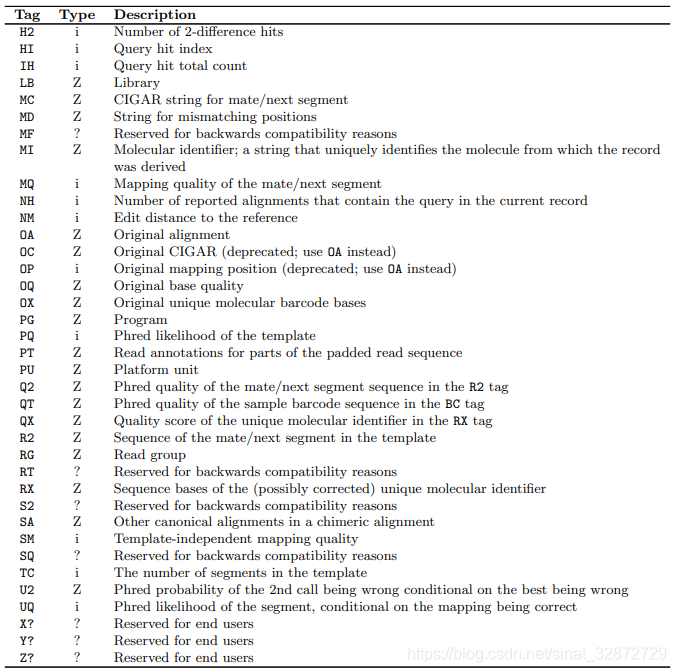
TAG:TYPE对应值VALUE的说明:TAG TYPE 说明 NM i i i 编辑距离。但是不包含头尾被剪切的序列。一般来说等于序列中error base的个数 MD Z Z Z 字符表示比对错配的位置信息 AS i i i 由比对工具生成的 比对得分 XS i i i 第二好的匹配的得分? RG Z Z Z Read group, 比对工具指定的信息,与sam文件的header @RG的ID相同 SA Z Z Z 候选的比对位置,相当于这条reads可比对到ref的多个位置 MC Z Z Z 两端测序比对时,该read对应的pair read的CIGAR值 XA Z Z Z 更多的候选比对位置? AM i i i 最小比对质量值? YS i i i mate 序列匹配的得分? XN i i i 在参考序列上模糊碱基的个数? XM i i i 错配的个数? XO i i i gap open的个数,针对于比对中的插入和缺失? XG i i i gap 延伸的个数,针对于比对中的插入和缺失 YF i i i 该reads被过滤掉的原因。可能为LN(错配数太多,待查证)、NS(read中包含N或者.)、SC(match bonus低于设定的阈值)、QC(failing quality control,待证) YT Z Z Z 值为UU表示不是pair中一部分(单末端?)、CP(是pair且可以完美匹配)、DP(是pair但不能很好的匹配)、UP(是pair但是无法比对到参考序列上) MD Z Z Z 比对上的错配碱基的字符串表示 
NM: 跟参考序列比,错配/插入/缺失的碱基数。 - 对应模糊碱基,比如:N(一般指A/T/C/G其中1个)、R(一般A/G其中一个)等,仍在统计时视为错配; - “=”碱基(目前没见过)视为匹配。CIGAR值为N/P/S/H时,不作为错配统计; - 插入和缺失统计时,每一个碱基视为一次错配; 注意:该值的定义可能在不同的比对工具之间存在差异。- 1
- 2
- 3
- 4
- 5

MD标签,Z: 字符串类型,正则匹配字符:`[0-9]+(([A-Z]|\^[A-Z]+)[0-9]+)*` 该字段目的在于,不看reference的情况下,进行SNP/indel的变异检测。 例如:`10A5^AC6`是指从比对到最左端ref的碱基,有10个匹配上,后面有一个A碱基在ref上与比对的序列不同,接着又5个碱基比对到ref, 后面有2bp的碱基AC发生了deletion,最后又6个碱基比对到ref。MD字段应该与CIGAR字符相匹配。 字符说明: `数字`:完全相同匹配的碱基数; `A/T/C/G`: (连续碱基)前面除了数字是匹配碱基数,没有其他字符,表示mismatch(read碱基与ref不同); `^`表示 后面的碱基是deletion; 注意:如果发生insertion, 在MD标签中没有对应的信息。(CIGAR值对应insertion的符号是"I")- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
正则匹配问题? SAMtags
sam tag文档中对MD使用的正则匹配字符应该使用:
(在匹配出现,连续del时匹配用"+“, 连续错配时(或“|”符号前)没有用”+"匹配连续错配情形)
不过,一般连续的单碱基的错配比较少见,可能不考虑连续单个碱基的错配也可。([0-9]+)(([A-Z]+|\^[A-Z]+)[0-9]+)*- 1
SA: 另一个候选比对的位置。(有HardClip) 有hardclip时,对应的flag值:256 使用pysam判断的属性是`is_secondary`- 1
- 2
- 3

BWA给出的对应Tag的解释:bwa.1 man
bwa Web
samtools Web
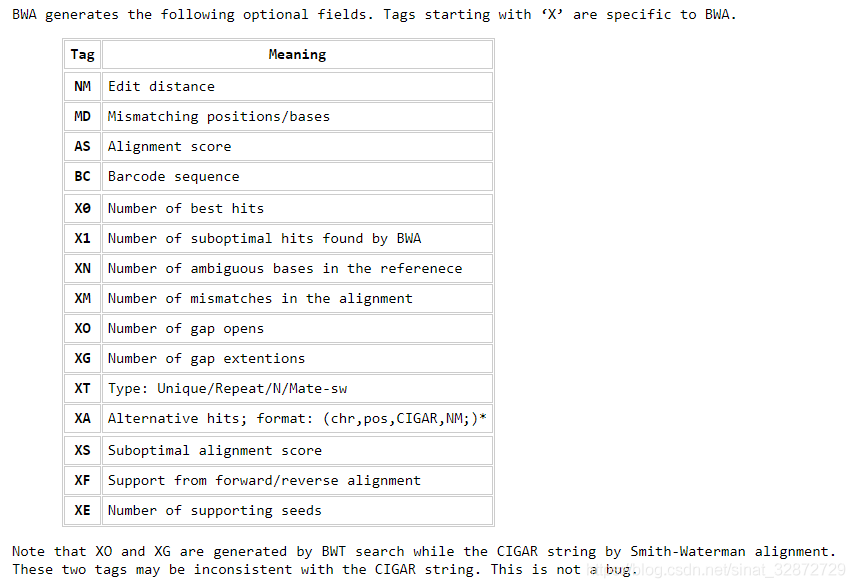
2021.5.12更新
CIGAR:值对应的字符,常用的pysam对应的数值:
CIGAR pysam 说明 M M M 0 matchd, 比对match(注意也有mismatch) I I I 1 insertion D D D 2 deletion N N N 3 skip,较长的deletion S S S 4 softclip H H H 5 hardcliip 问题:
bwa比对默认是把所有可能的比对结果都输出?指定参数输出比对次数。tag 查询:
https://www.samformat.info/sam-format-alignment-tags -
相关阅读:
【附源码】计算机毕业设计JAVA校园社团管理系统
JAVA学习------ConcurrentHashMap实现原理
java.lang.RuntimeException: java.security.InvalidKeyException: Illegal key size
MySQL8.0 索引优化-invisible index
微信小程序三种授权登录以及授权登录流程讲解
改进 hibernate-validator,新一代校验框架 validator 使用介绍 v0.4
matplotlibzzz颜色和双划线设置
git cherry pick
方块栈问题
linux并发服务器 —— linux网络编程(七)
- 原文地址:https://blog.csdn.net/sinat_32872729/article/details/104857278