-
R语言学习笔记
一.准备环境
下载R语言和Rtools,https://mirrors.tuna.tsinghua.edu.cn/CRAN/
下载Rstudio,https://posit.co/downloads/二.认识控制台
三.R包
R包并不是凭空产生的,都是作者编译好函数后封装成包,然后将其上传到某个平台储存。目前,R包主要都可以从这三大平台下载,分别是CRAN官网平台,Bioconductor生信平台及Github第三方平台。
简单介绍一下三个平台。
1.CRAN存储了R最新版本的代码和文档的服务器 。这是R语言官方平台,市面上常用的R包以及已经功能完整度比较高的R包基本都会上传到CRAN官方平台,因此通常可以从rstudio直接用代码下载。另外,可以从官网平台下载所需压缩包,然后在本地进行安装。
2.生物信息学领域的Bioconductor平台,它提供的R包主要为基因组数据分析和注释工具。这个平台主要上传了很多关于生信技术相关的R包,如果在遇到相关R包不能从官方CRAN平台下载时,可以优先考虑从这个平台下载。
3.面向开源及私有软件的第三方平台–Github。R包的作者更愿意将其存储在该平台,因此很多时候需要在上面下载 。如果前两个平台都无法下载,这时可以考虑第三方平台github,这里包含了99%的R包。#报错用英文显示,方便出错了取搜解决办法 Sys.setenv(LANGUAGE = "en") #安装CRAN下的包 install.packages("BiocManager") #可能会报错:构建R包需要Rtools,但当前未安装。在继续之前,请下载并安装相应版本的Rtools #加载BiocManager包 library(BiocManager) #安装Bioconductor下的包 BiocManager::install("GEOquery")- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
运行R包下的函数首先要加载R包
#加载GEOquery包 library(GEOquery) ##设置路径 getwd()#获取当前路径 setwd("E:\\Document\\RFiles") #用双斜杠 ?getGEO #查看函数参数 ##下载数据 getGPL = F 这个代表不下载平台注释文件,因为有时候网络不稳定。后面我们会在网页中下载,然后读取。 gset = getGEO('GSE12417', destdir=".",getGPL = F)- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
四.数据结构
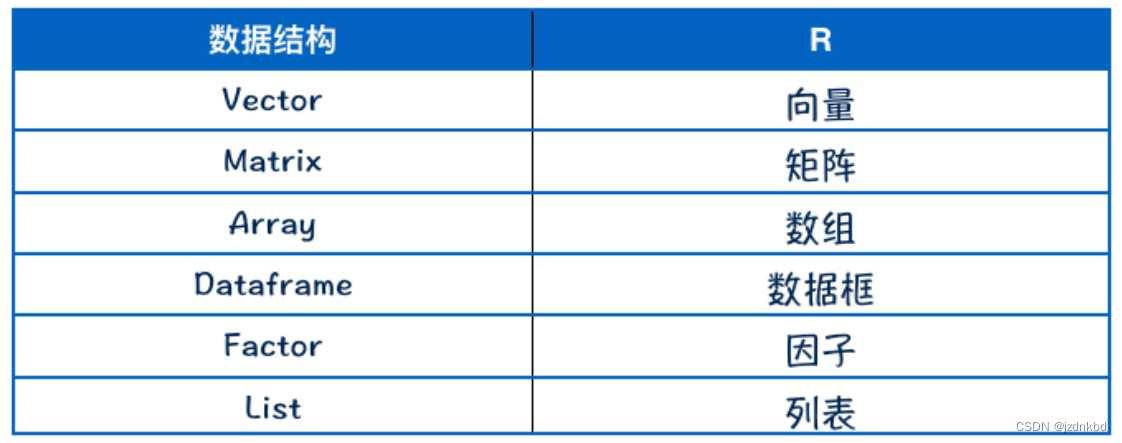
1.向量Vector
向量是用于存储数值型、字符型或逻辑型数据的一维数组。
两个特点 1.有序性 2.存储单一数据类型1.1创建向量
你需要认真学习以下函数:
c(): 用来建立向量的函数;seq(): 该函数用于创建包含from~end数值的向量;rep(): 该函数用来创建保存重复值的向量;length(): 用来计算向量长度的函数;names(): 该函数用来对向量各元素命名。我们可以用函数
c()来创建向量。比如:a <- c(1,2,3,4,6,7,9,0)#存储数值型 b <- c("哎","哟","喂啊")#存储字符型 c <- c(TRUE, FALSE, TRUE, TRUE)#存储逻辑型- 1
- 2
- 3
如果数据类型不同,会自动转化为同一类型
可以用mode()或class()函数查看对象数据类a <- c(7,TRUE);mode(a) #结果是"numeric" b <- c(7,"哎");mode(b) #结果是"character" c <- c(TRUE,"哎");mode(c) #结果是"character" d <- c(7,TRUE,"哎");mode(d) #结果是"character"- 1
- 2
- 3
- 4
无论什么类型的数据,缺失数据总是用
NA(不可用)来表示
如果想用键盘输入一些数据也是可以的,只需要直接使用默认选项的scan()函数:z <- scan()
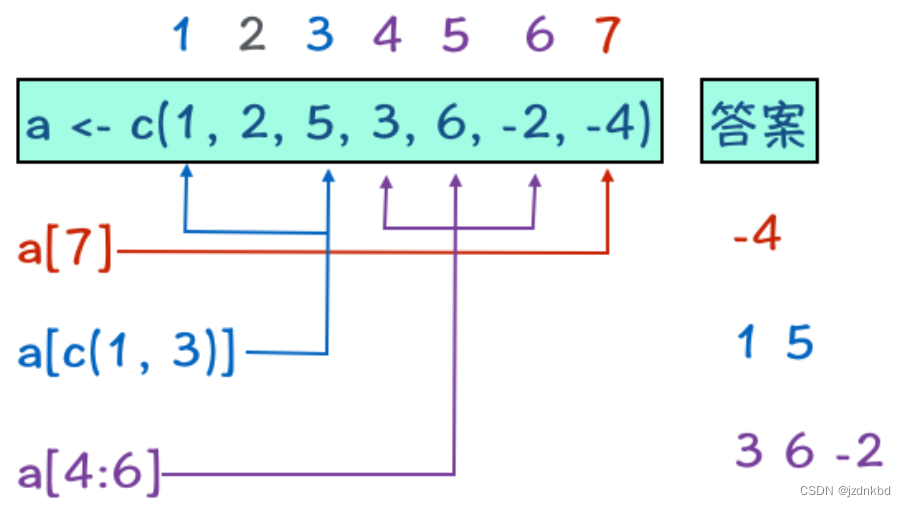
1.2访问向量中的数据
通过在
[ ]中给出元素所在位置的数值,我们就可以访问向量中的元素了。
有4种访问方式,看下图:

示例如下:
#删除第二个元素 a=a[-2]- 1
- 2
x[-1]表示将第一个元素删除。但是既然说的是负整数,如果非整数,比如x[-0.5]也不会报错,会默认删除第一个元素。x[-2.5]删除第二个元素。
空下标与零下标
x[ ]表示取x的全部元素作为子集。 这与x本身不同。
x[0]是一种少见的做法,结果返回类型相同、长度为零的向量,相当于空集。1.3向量的循环补齐
两个向量长度相等时,向量间的运算是相应位置的数字进行运算,运算后结果返回原位置。
但如果长度不相等呢?
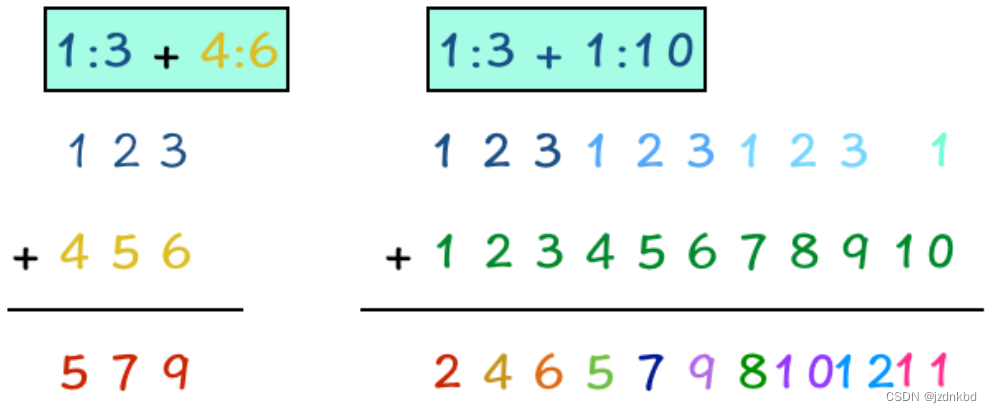
R会自动循环补齐,也就是它会自动重复较短的向量,直到与另外一个向量匹配
2.矩阵matrix
R中的矩阵与数学中的矩阵一样,由指定的行(row)与列(column)构成。
和向量一样,矩阵也只能保存同种数据类型的数据。2.1创建矩阵
你需要认真学习以下函数:
matrix(): 用来建立矩阵的函数;nrow():求矩阵的行数;ncol(): 求矩阵的列数;dim(): 求对象的维数;t(): 求矩阵的转置矩阵;solve(): 从方程a%*%x = b中求x。若不指定b, 则求a的逆矩阵。

x=matrix(c(1,3,4,2,-9,4,7,0,8),ncol=3) x- 1
- 2

给出的数据个数,要等于想要建立矩阵的元素个数才可以,否则就会报错
函数rownames()和colnames()可以为矩阵指定行名和列名。比如:rownames(x)=c("Lisa","Bob","Jay") colnames(x)=c("成绩1","成绩2","成绩3") x- 1
- 2
- 3
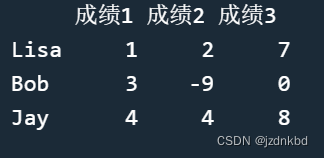
当没有行名时,我们用rownames()查看行名,会返回NULL
2.2访问矩阵中的数据
借助索引或行名与列名,就可以访问矩阵中的数据

与向量类似哟,如果索引是负数,则表示排除指定行或列;若索引为向量,则可以从矩阵中一次获取多个值。若想获取整行或整列,只要在指定行或列的位置上不写索引即可。#获取第一行、第三行与第1列、第三列数据的交集 x[-2,-2] #请使用行名、列名查出Bob的成绩2 x["Bob","成绩2"]- 1
- 2
- 3
- 4

矩阵与标量、矩阵与矩阵间的四则运算如下表所示:

除了简单的四则运算,矩阵还有特殊的运算需要函数来帮忙。
我们用
t()函数来求矩阵的转置矩阵;我们用
solve(a, b)函数来为方程 a %*% x = b求解。其中 a为矩阵,b为向量或矩阵;若不指定b,则求 a的逆矩阵。
我们用
nrow()函数来求矩阵的行数;我们用
ncol()函数来求矩阵的列数;我们用
dim()函数来求矩阵的维数,当然也可以用它来设置矩阵维数。比如:#将3*2的矩阵x改为2*3,并输出矩阵x x <- matrix(c(1,3,4,2,-9,4), ncol=2) x dim(x) <- c(2,3) x- 1
- 2
- 3
- 4
- 5
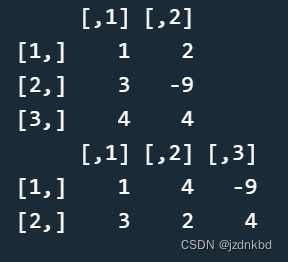
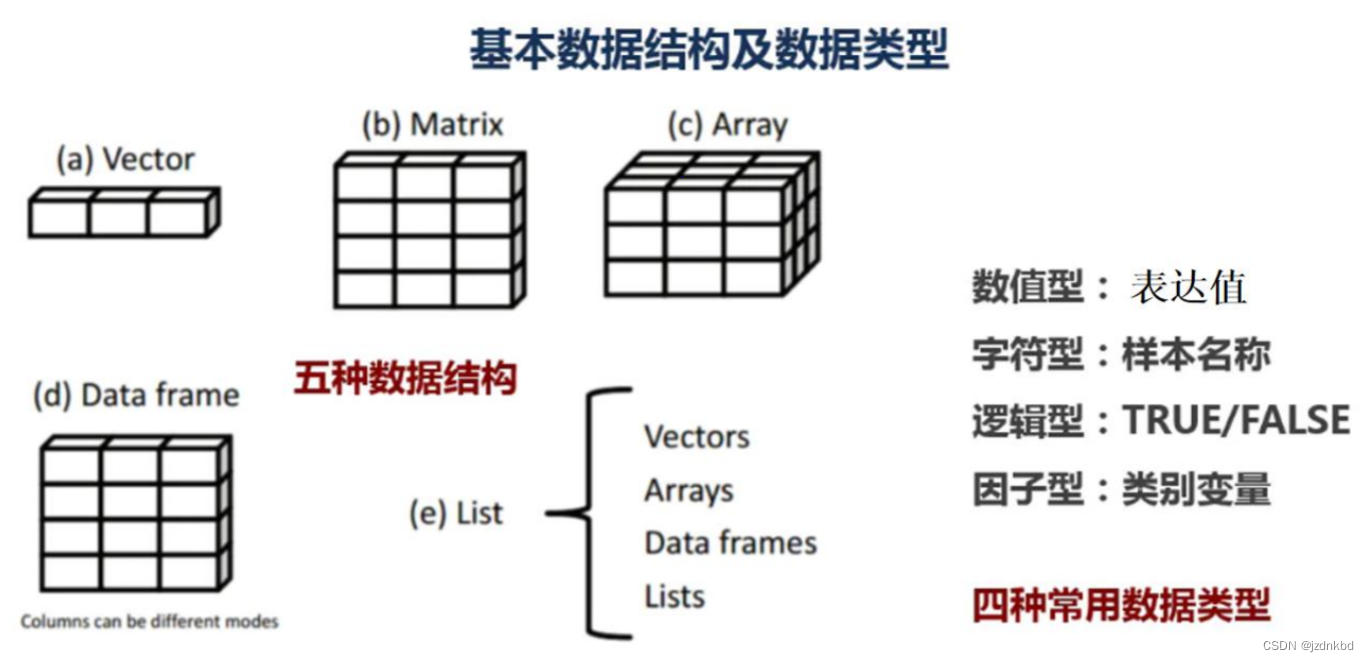
3数组Array
数组与矩阵类似,但是维度可以大于2。
比如,使用矩阵可以表现 2×3 维的数据,而使用数组则可以表现 2×3×4 维的数据。
和向量、矩阵一样,数组也只能保存同种数据类型的数据。3.1创建数组
你需要认真学习以下函数:
array(): 用来建立数组的函数;dim(): 求对象的维数。

array(1:12,dim=c(3,4))- 1

对于4×3×2维的数组,你们也可以理解为,它创造了2个 4×3的矩阵。x<-array(1:24,dim=c(4,3,2))- 1

3.2访问数组中的数据
访问数组中数据的方法和矩阵类似,因为他们只是维数不同而已。
所以,要想访问数组元素,我们一样可以使用索引、名称等进行访问。x[1, , ] x[ ,1, ] x[ , ,1] x[1,1, ] x[ ,1,1] x[1, ,1] x[1,1,1]- 1
- 2
- 3
- 4
- 5
- 6
- 7
4.数据框Dataframe
4.1创建数据框
你需要认真学习以下函数:
data.frame(): 用来建立数据框的函数;str(): 查看数据框结构。由于不同的列可以包含不同模式(数值型、字符型)的数据,数据框的概念跟矩阵相比更为一般。
数据框是R中最常处理的数据结构。
数据框可以通过函数 data.frame(col1, col2, col3, …) 来创建。
其中列向量col1,col2,col3可为任何类型。
每一列数据的模式必须唯一,不过你却可以将多个模式的不同列放到一起组成数据框。
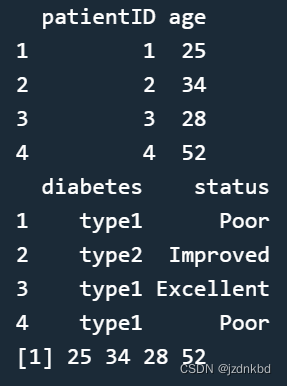
比如执行以下代码后:patientID <- c(1, 2, 3, 4) age <- c(25, 34, 28, 52) diabetes <- c("type1", "type2", "type1", "type1") status <- c("Poor", "Improved", "Excellent", "Poor") patientdata <- data.frame(patientID, age, diabetes, status) patientdata- 1
- 2
- 3
- 4
- 5
- 6
我们能得到结果:

4.2访问数据框中的数据
我们可以像之前一样使用索引来访问数据框中的元素,也可以用数据框特有的新办法来访问,那就是 $ 符号。
patientdata[1:2] patientdata[c("diabetes", "status")] patientdata$age- 1
- 2
- 3
下面是运行结果:
函数str()可以查看数据框结构str(patientdata)- 1
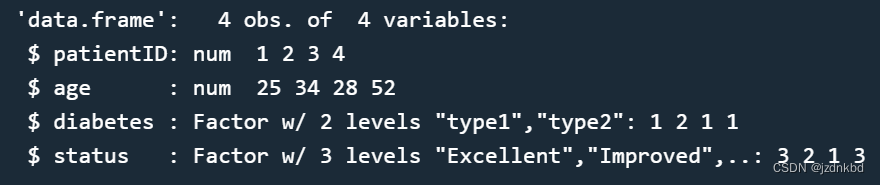 可以看到diabets和status是因子类型,下面会讲。
可以看到diabets和status是因子类型,下面会讲。names()函数用于返回数据框点的列名。使用 %in%运算符与names()函数,能够快速选取特定列。
patientdata[ ,names(patientdata)%in%c("patientID","diabetes")]- 1

反之~使用 ! 运算符可以排除特定列patientdata[ ,!names(patientdata)%in%c("patientID","diabetes")]- 1
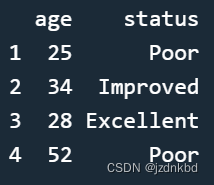
如果只访问一列,会返回向量,想避免这种类型转换的话,只需要设置drop=FALSE即可patientdata[ ,2] patientdata[ ,2,drop=F]- 1
- 2

5因子Factor
因子,简单来说,在 R 中,因子就是分类数据。
比如说,狗狗可以分类成“大型犬”“中型犬”“小型犬”,那“狗狗”就是顺序型的因子,它有3种水平(level),分别是“大型犬”“中型犬”“小型犬”。
那还有一些因子其实是无法比较大小的,我们叫它名义型的因子,比如政治倾向中的2种level,“左派”和“右派”。
5.1创建因子
你需要认真学习以下函数:
factor(): 用来建立因子的函数;nlevels(): 返回因子中的水平个数;levels(): 返回因子水平目录;ordered(): 创建有序因子。

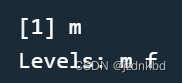
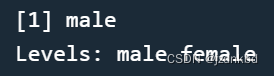
性别属于名义型数据,有“m”(male,男性)与“f”(female,女性)两种可能的值。下面的例子中,创建了性别的因子,保存了男性的变量在sex中。sex <- factor("m", c("m","f")) sex- 1
- 2
5.2访问因子中的数据
函数levels()可以获得因子水平的名称。
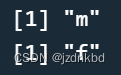
levels()返回的是向量,所以我们可以用索引获得各水平值。levels(sex)[1] levels(sex)[2]- 1
- 2
我们还可以修改因子变量中的水平值
还是用levels()函数
比如:把“m”修改为“male”,“f"修改为"female”levels(sex)<-c("male","female") sex- 1
- 2
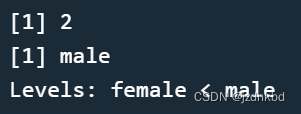
我们用nlevels()函数来获取因子水平个数;我们用ordered()函数来设置水平值的顺序。
nlevels(sex) ordered(sex,c("female","male")- 1
- 2
6.列表List
列表(list)是R的数据类型中最复杂的一种。它什么都存
某个列表中可能是若干向量、矩阵、数据框,甚至其他列表的组合。6.1创建列表
R中的列表与其他语言中的散列表(Hash table)或字典(Dictionary)非常类似,就是说,列表是以“(键,值)”对的形式保存数据的关联数组(Associative Array)。

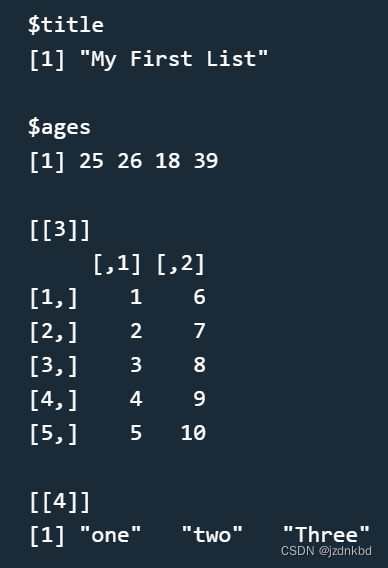
比如:创建一个列表,该列表有4个对象,分别是g、h、j、k
其中对象 g 的 name 为 title,对象 h 的 name 为 ages,其他对象没有 name
g 为 标量字符串“My First List”
h 为数值向量,包含元素 25,26,18,39
j 为 5*2 的矩阵,元素为1:10
k 为字符串向量,包含元素 “one”,“two”,“Three”x=list(title="My First List",ages=c(25,26,18,39),matrix(1:10,nrow=5),c("one","two","Three")) x- 1
- 2
6.2访问列表中的数据
访问列表中的数据,我们既可以使用索引又可以使用对象名。

输出列表时,其中的每个对象都会以 $name 的形式罗列出来。使用 x $name形式可以访问对象的数据。
x[n]返回的是(name, value)的子列表,不是value!
而x[[n]]返回的是对象的value!#输出该列表的第二个对象的 value x[[2]] #输出该列表第三个对象 x[2] #输出“ages”的value x$ages- 1
- 2
- 3
- 4
- 5
- 6

可以看到x[[[2]]]与x$ages结果一样五.获取表达矩阵
library(GEOquery) ##下载数据 getGPL = F 这个代表不下载平台注释文件,因为有时候网络不稳定。后面我们会在网页中下载,然后读取。 gset = getGEO('GSE12417', destdir=".",getGPL = F)- 1
- 2
- 3
View(gset)- 1
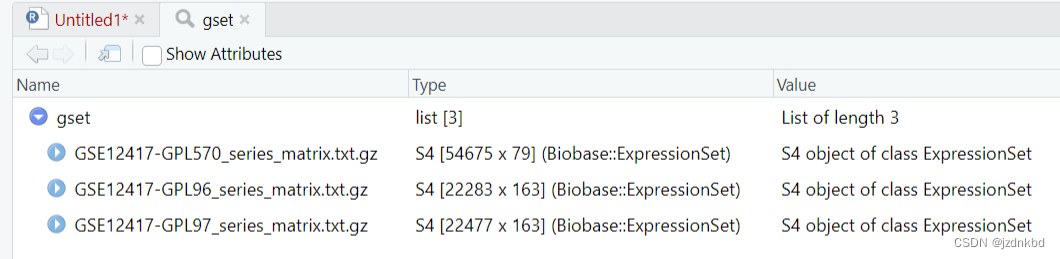
class(gset)- 1
class(gset)
[[1]] “list”可以看到gest对象是list类型数据,提取list类型数据用
[[ ]]#按序号提取GPL96 e2<-gest[[2]] #按名字提取 e2<-gset[["GSE12417-GPL96_series_matrix.txt.gz"]]- 1
- 2
- 3
- 4
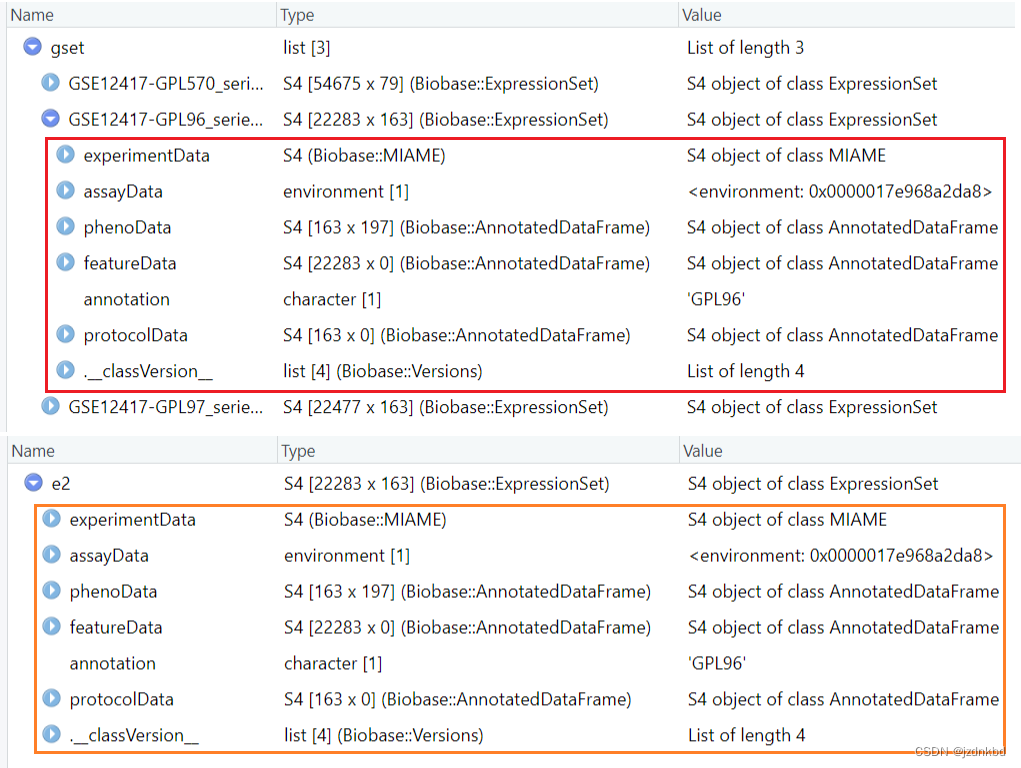
# s3:matrix 矩阵 data.frame() 数据框 character() 向量字符型 list 列表 # s4更复杂- 1
- 2
s4对象提取
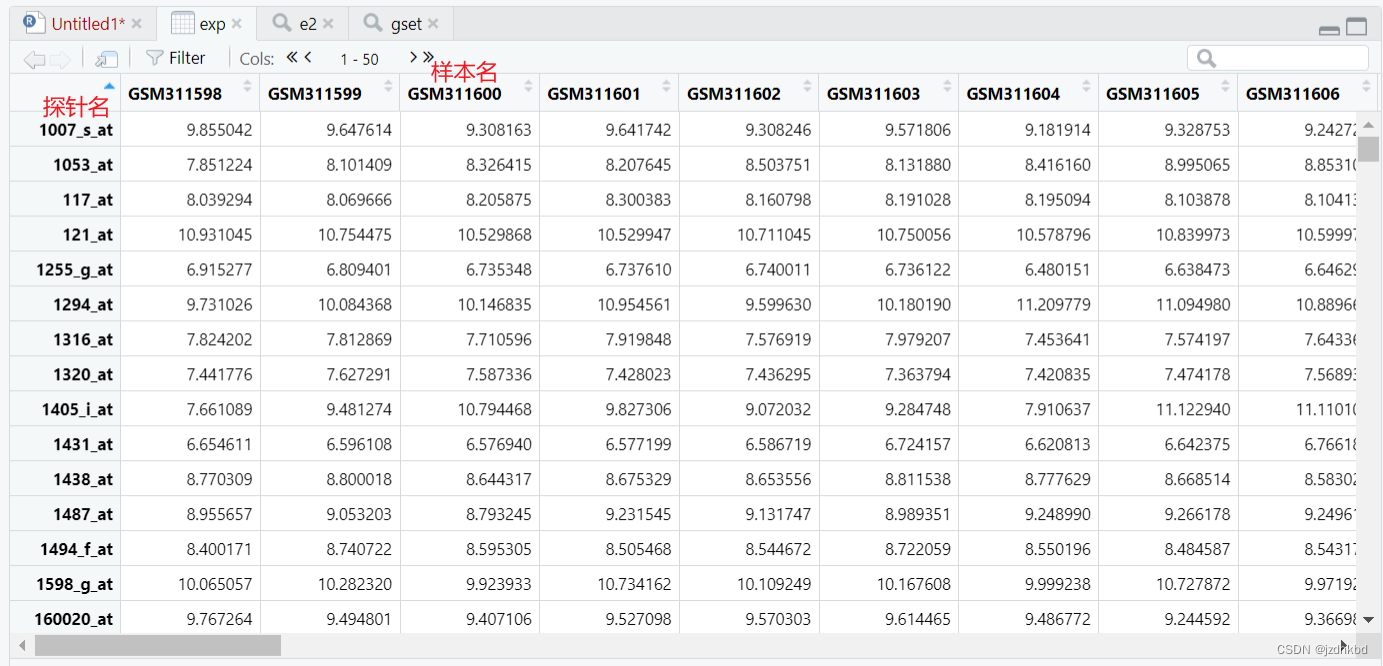
###提取表达矩阵 exp=exprs(e2) ##当然也可以用环境中的白色括号提取 exp2=e2@assayData[["exprs"]] class(exp) #[1] "matrix" "array" View(exp) ###用 @ 或 $符号 phe=e2@phenoData@data phel=e2@phenoData@varMetadata$labelDescription- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
环境对象保存save(gest,file="gset.rdata") #把gset对象保存为rdata数据文件 load("gset.rdata") #将这个对象文件加载到环境中 ####或者用下面一对函数保存为rds文件 saveRDS(gset,file="gse12417.rds") readRDS("gse12417.rds")- 1
- 2
- 3
- 4
- 5
整理探针转化文件
#e2<-gset[["GSE12417-GPL96_series_matrix.txt.gz"]] #e2是GPL96平台,首先下载文件- 1
- 2
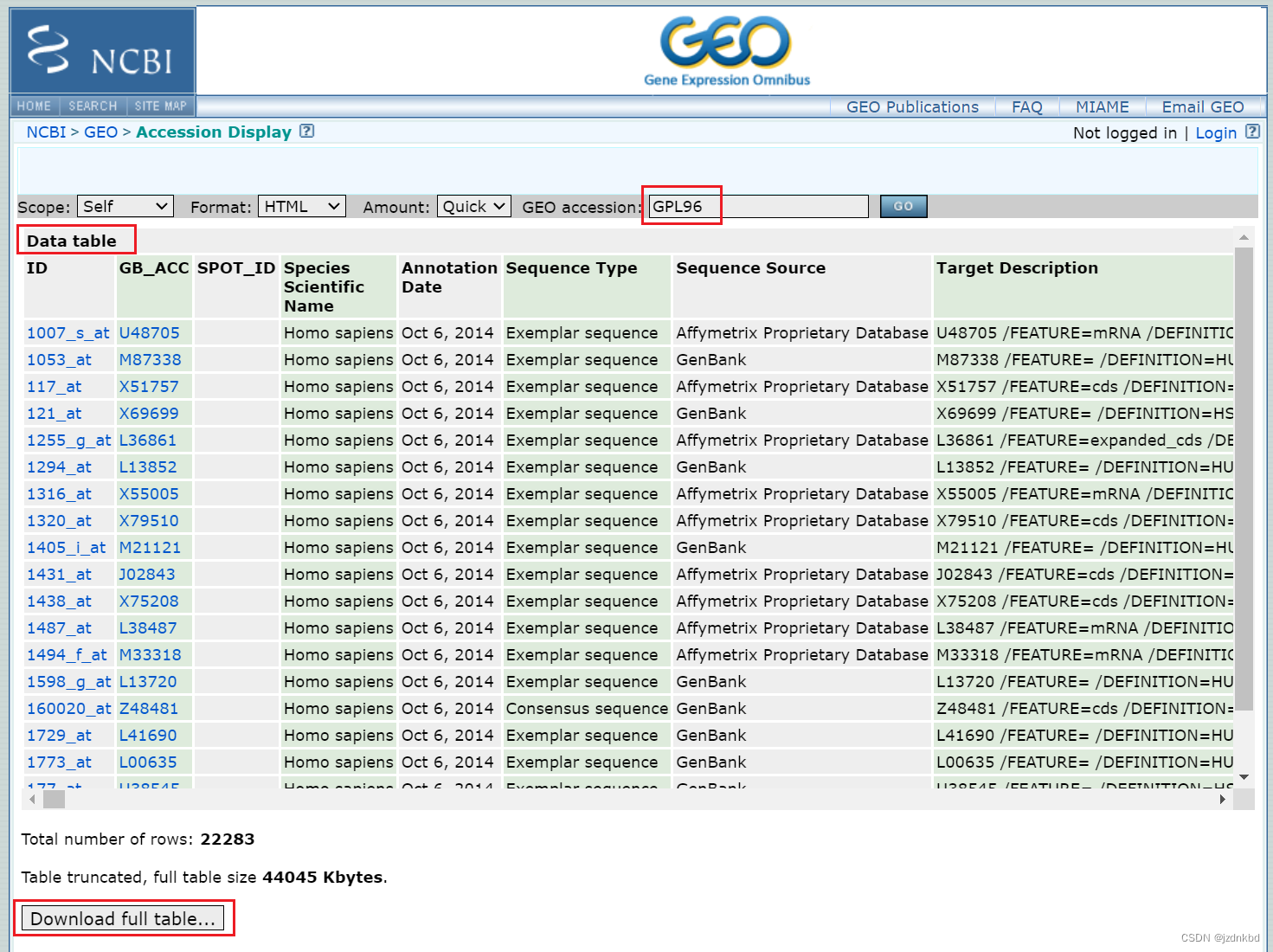
往右拉可以看到探针对应的基因名
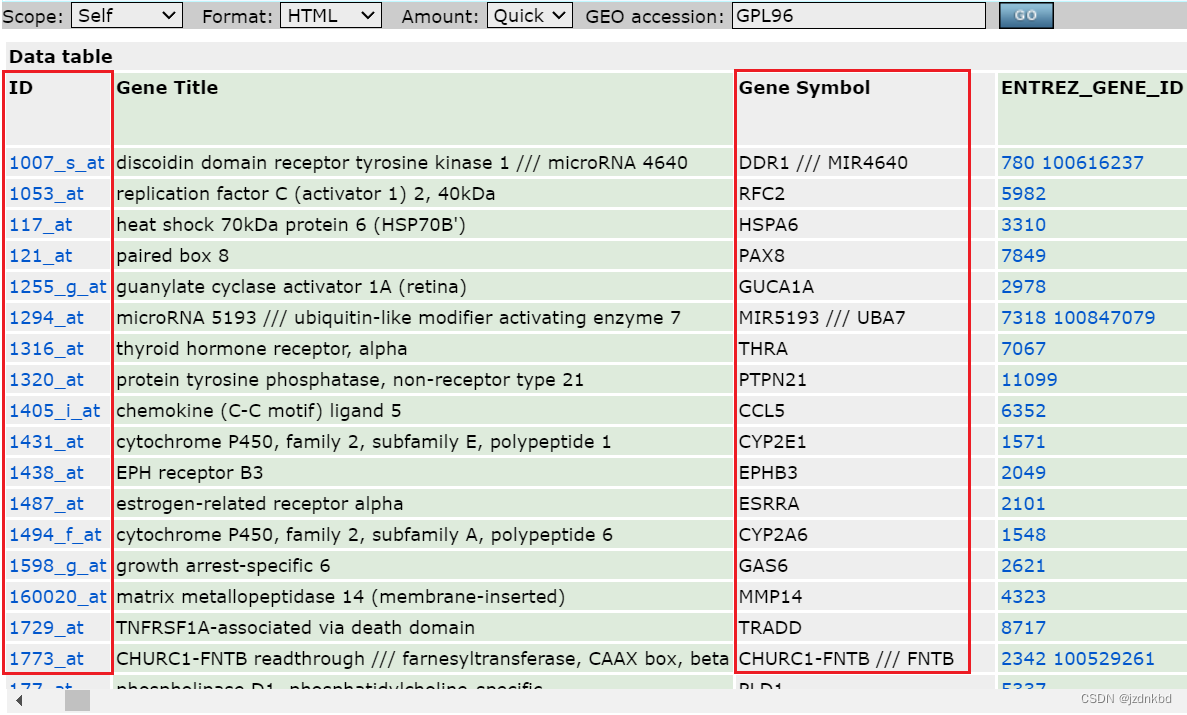
library(data.table) #用里面 fread 函数读取txt文件 anno=fread("GPL96-57554.txt",header = T,data.table = F) #用read,table读取 anno3=read.table("GPL96-57554.txt",sep="\t",header=T,fill=T) class(anno)#[1] "data.frame" View(anno)- 1
- 2
- 3
- 4
- 5
- 6
- 7
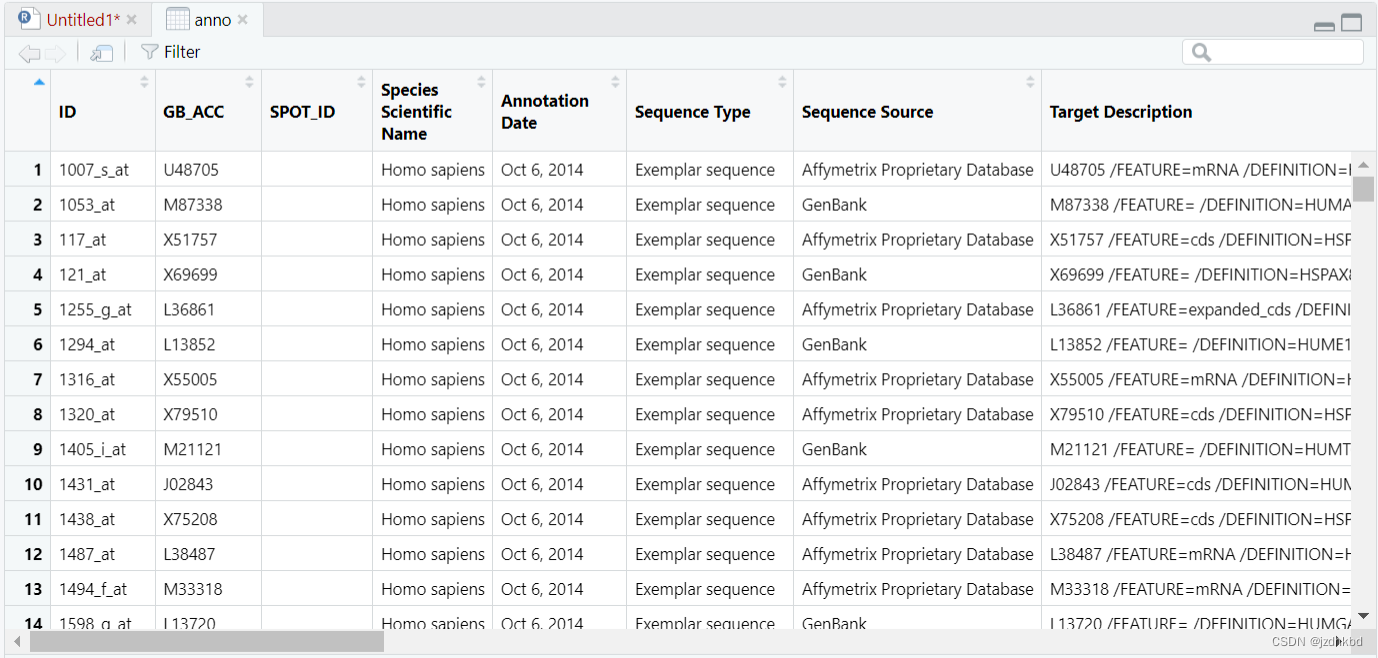
x1=colnames(anno) x2=rownames(anno)- 1
- 2
未完待续。。。。
-
相关阅读:
尝试用Unity还原蔚蓝(Celeste)—— 真·操控、移动、手感篇
技术分享 LINUX卸载oracle
鸿蒙HarmonyOS实战-ArkUI组件(Radio)
iframe内的通信(桥接方法),使用postMessage和使用自定义事件
爬虫获取页面源码
ubuntu18.04环境下部署ROS2 docker镜像全步骤
多路查找树
网页开发从无到有——html前端学习(七)
Python初级练习小实例(1-20例),1个实例多个例子相互参考
关于addEventListener的使用和注意项
- 原文地址:https://blog.csdn.net/Linux518/article/details/132788148