-
新版TCGAbiolinks包学习02:提取新版TCGA表达矩阵(tpm/count/fpkm)
现在使用
TCGAbiolinks下载转录组数据后,直接是一个SummarizedExperiment对象,这个对象非常重要且好用。因为里面直接包含了表达矩阵、样本信息、基因信息,可以非常方便的通过内置函数直接提取想要的数据,再也不用手扒了!!这个对象的结构是这样的:

是不是感觉和单细胞的
SingCellExperiment对象非常像~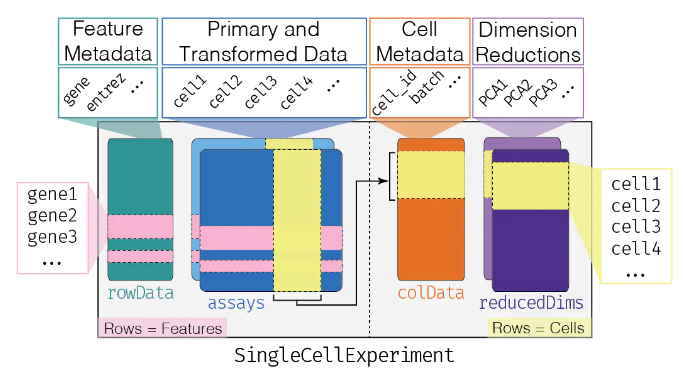
上次我们下载了常见的组学数据,今天学习下怎么提取数据,就以TCGA-READ的转录组数据为例。
分别提取mRNA和lncRNA的表达矩阵,还要添加gene symbol的那种!
加载数据和R包
加载之前下载好的数据。
rm(list = ls()) library(SummarizedExperiment) ## Loading required package: MatrixGenerics ## Loading required package: matrixStats ## ## Attaching package: 'MatrixGenerics' ## The following objects are masked from 'package:matrixStats': ## ## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse, ## colCounts, colCummaxs, colCummins, colCumprods, colCumsums, ## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs, ## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats, ## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds, ## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads, ## colWeightedMeans, colWeightedMedians, colWeightedSds, ## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet, ## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods, ## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps, ## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins, ## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks, ## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars, ## rowWeightedMads, rowWeightedMeans, rowWeightedMedians, ## rowWeightedSds, rowWeightedVars ## Loading required package: GenomicRanges ## Loading required package: stats4 ## Loading required package: BiocGenerics ## ## Attaching package: 'BiocGenerics' ## The following objects are masked from 'package:stats': ## ## IQR, mad, sd, var, xtabs ## The following objects are masked from 'package:base': ## ## anyDuplicated, append, as.data.frame, basename, cbind, colnames, ## dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep, ## grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget, ## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank, ## rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply, ## union, unique, unsplit, which.max, which.min ## Loading required package: S4Vectors ## ## Attaching package: 'S4Vectors' ## The following objects are masked from 'package:base': ## ## expand.grid, I, unname ## Loading required package: IRanges ## ## Attaching package: 'IRanges' ## The following object is masked from 'package:grDevices': ## ## windows ## Loading required package: GenomeInfoDb ## Loading required package: Biobase ## Welcome to Bioconductor ## ## Vignettes contain introductory material; view with ## 'browseVignettes()'. To cite Bioconductor, see ## 'citation("Biobase")', and for packages 'citation("pkgname")'. ## ## Attaching package: 'Biobase' ## The following object is masked from 'package:MatrixGenerics': ## ## rowMedians ## The following objects are masked from 'package:matrixStats': ## ## anyMissing, rowMedians load("TCGA-mRNA/TCGA-READ_mRNA.Rdata") se <- data- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
- 58
- 59
- 60
- 61
- 62
- 63
- 64
- 65
- 66
- 67
- 68
- 69
- 70
- 71
这个
se就是你的对象,含有coldata, rowdata, meta-data,以及最重要的assay,共有6个assay探索SummarizedExperiment对象
se ## class: RangedSummarizedExperiment ## dim: 60660 177 ## metadata(1): data_release ## assays(6): unstranded stranded_first ... fpkm_unstrand fpkm_uq_unstrand ## rownames(60660): ENSG00000000003.15 ENSG00000000005.6 ... ## ENSG00000288674.1 ENSG00000288675.1 ## rowData names(10): source type ... hgnc_id havana_gene ## colnames(177): TCGA-AG-3580-01A-01R-0821-07 ## TCGA-AF-2692-11A-01R-A32Z-07 ... TCGA-AG-3894-01A-01R-1119-07 ## TCGA-AG-3574-01A-01R-0821-07 ## colData names(107): barcode patient ... paper_vascular_invasion_present ## paper_vital_status- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
看看这个对象,它告诉你:
- 类型是:RangedSummarizedExperiment
- 维度:60660行,177列
- 6个assay以及它们的名字
- 表达矩阵的行名
- 行信息(也就是基因信息),比如gene id,gene name,gene type
- 表达矩阵的列名(也就是样本名)
- 列信息,也就是样本信息,比如生存时间、生存状态这些
太齐全了有没有!!
每个assay你可以理解为一个表达矩阵,我们需要的counts矩阵、TPM矩阵、FPKM矩阵就是其中一个~
# 查看每个assay的名字 names(assays(se)) ## [1] "unstranded" "stranded_first" "stranded_second" "tpm_unstrand" ## [5] "fpkm_unstrand" "fpkm_uq_unstrand"- 1
- 2
- 3
- 4
每个基因属于mRNA还是lncRNA存储在
rowData中,这个rowData你可以理解为一个包含基因各种信息的数据框。其中
gene_type是基因类型,帮助我们区分到底是lncRNA还是mRNA,当然还包括很多其他类型。# 提取rowData rowdata <- rowData(se) # 看看rowData包括哪些内容,可以看到里面有我们需要的gene_name和gene_type names(rowdata) ## [1] "source" "type" "score" "phase" "gene_id" ## [6] "gene_type" "gene_name" "level" "hgnc_id" "havana_gene" # gene_type是基因类型,看看有哪些 table(rowdata$gene_type) ## ## IG_C_gene IG_C_pseudogene ## 14 9 ## IG_D_gene IG_J_gene ## 37 18 ## IG_J_pseudogene IG_pseudogene ## 3 1 ## IG_V_gene IG_V_pseudogene ## 145 187 ## lncRNA miRNA ## 16901 1881 ## misc_RNA Mt_rRNA ## 2212 2 ## Mt_tRNA polymorphic_pseudogene ## 22 48 ## processed_pseudogene protein_coding ## 10167 19962 ## pseudogene ribozyme ## 18 8 ## rRNA rRNA_pseudogene ## 47 497 ## scaRNA scRNA ## 49 1 ## snoRNA snRNA ## 943 1901 ## sRNA TEC ## 5 1057 ## TR_C_gene TR_D_gene ## 6 4 ## TR_J_gene TR_J_pseudogene ## 79 4 ## TR_V_gene TR_V_pseudogene ## 106 33 ## transcribed_processed_pseudogene transcribed_unitary_pseudogene ## 500 138 ## transcribed_unprocessed_pseudogene translated_processed_pseudogene ## 939 2 ## translated_unprocessed_pseudogene unitary_pseudogene ## 1 98 ## unprocessed_pseudogene vault_RNA ## 2614 1- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
gene_name就是gene_symbol,我们的id转换就用这一列信息。# gene_name就是gene_symbol,就是我们需要的 head(rowdata$gene_name) ## [1] "TSPAN6" "TNMD" "DPM1" "SCYL3" "C1orf112" "FGR" length(rowdata$gene_name) ## [1] 60660- 1
- 2
- 3
- 4
- 5
还有一个重要的知识点是:
SummarizedExperiment对象可以取子集,就像对数据框取子集那样,选择符合条件的行和列,并且子集也是SummarizedExperiment对象!rowdata <- rowData(se) # 分别提取mRNA和lncRNA的SummarizedExperiment对象 # 根据gene_type取子集,太简单了! se_mrna <- se[rowdata$gene_type == "protein_coding",] se_lnc <- se[rowdata$gene_type == "lncRNA"] se_mrna #还是一个SummarizedExperiment对象,神奇! ## class: RangedSummarizedExperiment ## dim: 19962 177 ## metadata(1): data_release ## assays(6): unstranded stranded_first ... fpkm_unstrand fpkm_uq_unstrand ## rownames(19962): ENSG00000000003.15 ENSG00000000005.6 ... ## ENSG00000288674.1 ENSG00000288675.1 ## rowData names(10): source type ... hgnc_id havana_gene ## colnames(177): TCGA-AG-3580-01A-01R-0821-07 ## TCGA-AF-2692-11A-01R-A32Z-07 ... TCGA-AG-3894-01A-01R-1119-07 ## TCGA-AG-3574-01A-01R-0821-07 ## colData names(107): barcode patient ... paper_vascular_invasion_present ## paper_vital_status se_lnc ## class: RangedSummarizedExperiment ## dim: 16901 177 ## metadata(1): data_release ## assays(6): unstranded stranded_first ... fpkm_unstrand fpkm_uq_unstrand ## rownames(16901): ENSG00000082929.8 ENSG00000083622.8 ... ## ENSG00000288667.1 ENSG00000288670.1 ## rowData names(10): source type ... hgnc_id havana_gene ## colnames(177): TCGA-AG-3580-01A-01R-0821-07 ## TCGA-AF-2692-11A-01R-A32Z-07 ... TCGA-AG-3894-01A-01R-1119-07 ## TCGA-AG-3574-01A-01R-0821-07 ## colData names(107): barcode patient ... paper_vascular_invasion_present ## paper_vital_status- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
提取表达矩阵
有了这些东西,就可以提取表达矩阵了,直接使用
assay()搞定!# mRNA的counts矩阵 expr_counts_mrna <- assay(se_mrna,"unstranded") # mRNA的tpm矩阵 expr_tpm_mrna <- assay(se_mrna,"tpm_unstrand") # mRNA的fpkm矩阵 expr_fpkm_mrna <- assay(se_mrna,"fpkm_unstrand") # lncRNA的counts矩阵 expr_counts_lnc <- assay(se_lnc,"unstranded") # lncRNA的tpm矩阵 expr_tpm_lnc <- assay(se_lnc,"tpm_unstrand") # lncRNA的fpkm矩阵 expr_fpkm_lnc <- assay(se_lnc,"fpkm_unstrand")- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
简单!方便!快捷!
随便展示下:
expr_counts_mrna[1:10,1:2] ## TCGA-AG-3580-01A-01R-0821-07 TCGA-AF-2692-11A-01R-A32Z-07 ## ENSG00000000003.15 3199 5839 ## ENSG00000000005.6 6 91 ## ENSG00000000419.13 828 1867 ## ENSG00000000457.14 386 639 ## ENSG00000000460.17 228 289 ## ENSG00000000938.13 130 452 ## ENSG00000000971.16 277 5170 ## ENSG00000001036.14 1648 2946 ## ENSG00000001084.13 823 2414 ## ENSG00000001167.14 619 1487- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12

是不是很简单?肯定比之前简单多了吧?
添加gene_symbol
添加
gene_symbol也就非常简单了,只要提取gene_name这一列,然后和原来的表达矩阵合并即可!# 先提取gene_name symbol_mrna <- rowData(se_mrna)$gene_name head(symbol_mrna) ## [1] "TSPAN6" "TNMD" "DPM1" "SCYL3" "C1orf112" "FGR" symbol_lnc <- rowData(se_lnc)$gene_name head(symbol_lnc) ## [1] "LINC01587" "AC000061.1" "AC016026.1" "IGF2-AS" "RRN3P2" ## [6] "AC087235.1"- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
和你喜欢的表达矩阵合并就行了:
expr_counts_mrna_symbol <- cbind(data.frame(symbol_mrna), as.data.frame(expr_counts_mrna))- 1
- 2

非常顺利~
但是呢,此时gene_symbol是有重复的,看上图中就有2个CD99,需要去重!
去重复也很简单,这里我们保留最大的那个。
suppressPackageStartupMessages(library(tidyverse)) expr_read <- expr_counts_mrna_symbol %>% as_tibble() %>% # tibble不支持row name,我竟然才发现! mutate(meanrow = rowMeans(.[,-1]), .before=2) %>% arrange(desc(meanrow)) %>% distinct(symbol_mrna,.keep_all=T) %>% select(-meanrow) %>% column_to_rownames(var = "symbol_mrna") %>% as.data.frame()- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
不过还是要注意,gene_symbol是有重复的,需要去重复哦~
结果就变成大家最熟悉的表达矩阵了:

这样一个表达矩阵就搞定了!
本文由mdnice多平台发布
-
相关阅读:
【Redis】面试常见问题
【图像分类】【深度学习】【Pytorch版本】AlexNet模型算法详解
AWK用法全解与sed去掉sql最后一个字段哪一行的逗号
和数集团海外团队受邀参加俄罗斯2022区块链峰会
LVGL双向链表学习笔记
自行车轴承市场调研:预计2028年将达到25.6亿美元
xilinx fpga ultrascale 器件GTX参考时钟注意点
LCP 02.分式化简
CSDN 重新开放付费资源的上传了,但要求如下
视觉任务相关的问题汇总
- 原文地址:https://blog.csdn.net/Ayue0616/article/details/126452387
