-
MindSpore社区群组介绍系列之三——WG- Molecular Modeling


分子动力学[0]最早在20世纪50年代由物理学家提出,是一套分子模拟方法,该方法主要是依靠计算机来模拟分子、原子体系的运动,是一种多体模拟方法。
分子动力学如今被广泛应用于广泛应用于物理、化学、生物、材料、医学等领域:
-
通过研究石墨烯中的离子输运来了解石墨烯的机械性质,从而为创造新材料做好基础;
-
在新冠肺炎病毒变异研究中利用分子动力学来预测药物靶位,从而研制出高效的疫苗。
可以说是当之无愧的基础核心技术。
具体来说分子动力学是通过对分子、原子在一定时间内运动状态的模拟,从而以动态观点考察系统随时间演化的行为。通常分子、原子的轨迹是通过数值求解牛顿运动方程得到,势能(或其对笛卡尔坐标的一阶偏导数,即力)通常可以由分子间相互作用势能函数、分子力学力场、全始计算给出。
势能函数的计算方式通常有两种:
-
量子力学:
又称为“从头算”(ab initio)方法或“第一性原理”( First Principle )方法,指基于量子力学基本原理,直接求解薛定谔方程(Schrödinger Equation )的量子化学计算方法。其特点是没有经验参数,精度高,可以描述化学键的断裂和生成等涉及电子结构变化的过程,但计算耗时极长。
-
分子力学:
使用分子力场(Force Field)来描述体系的势能函数。传统的分子力场使用一系列经验性的数学函数,对用QM放法计算或者实验得到的数据进行参数拟合,从而得到体系的势能函数。该方法的特点是计算速度快,但精度受限于所拟合的对象,可迁移性差,且通常无法模拟化学键的断裂和生成,也即无法模拟化学反应的过程。

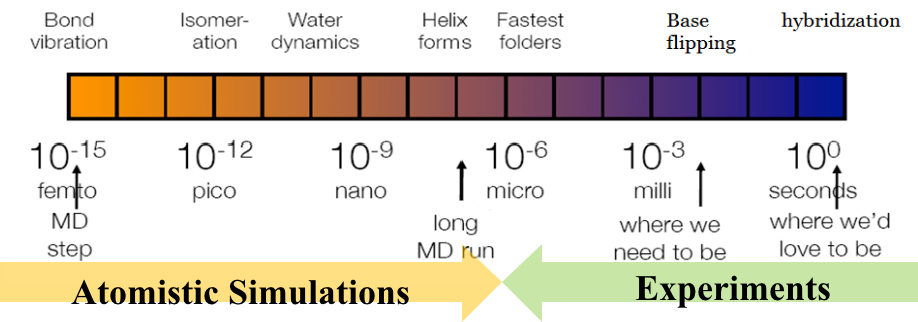
优化的对象→MD的可达到的时间尺度:
能量函数的计算复杂度
积分步长
体系粒子数
优化的目标→如何拓展MD的时间尺度:
特殊硬件: ×10^3
增强抽样方法:×10^3~6
能够快速计算的替代势能函数(light-speed surrogate energy function):×10^3~6

问题:
-
多年前开始使用C/C++或Fortran编写,历史包袱沉重
-
需要对所有函数手工写其对应的导数,以计算势能和力
-
需要手工处理并行(MPI)操作
-
在专用加速设备(如GPU)上运行需要单独编写对应代码,不同设备之间的迁移非常麻烦
-
传统分子力场模型存在“精度”和“速度”之间的矛盾
应对策略:
-
使用新的库重新编写MD软件
-
使用自动微分技术处理势能和力
-
使用自动并行技术实现MD并行操作
-
使用TVM等技术实现代码在不同设备间的迁移
-
使用深度分子模型替代传统分子力场模型

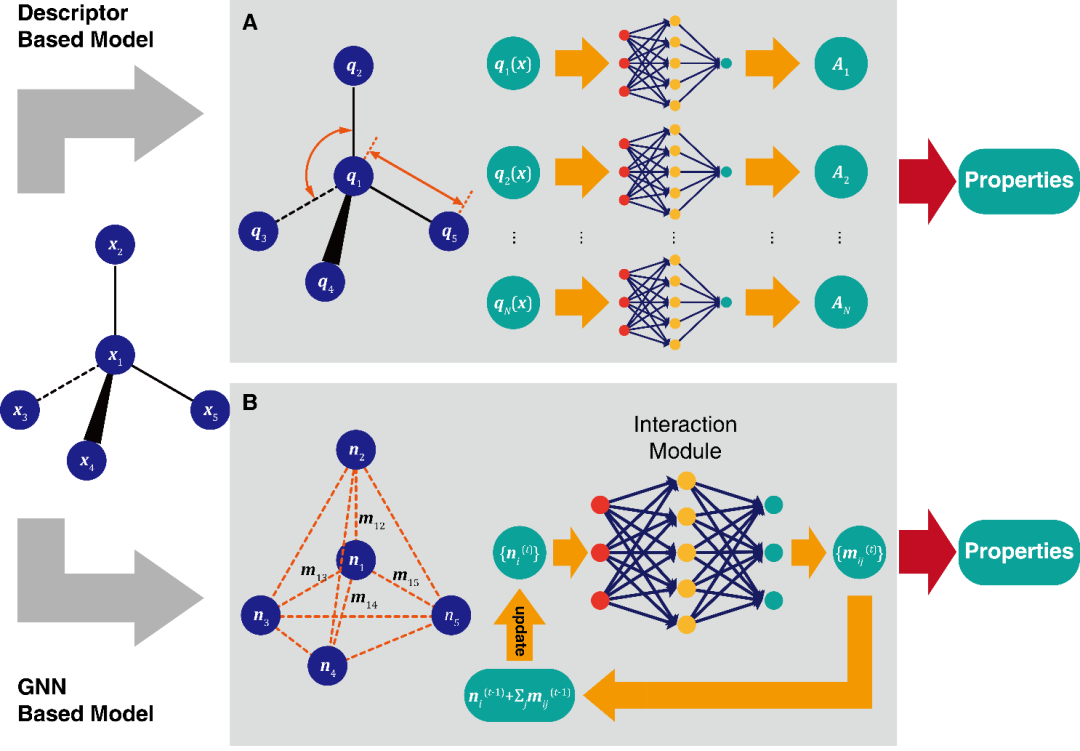
现有的深度分子模型架构:
基于描述符(Descriptor)的模型
基于图神经网络(Graph Neural Network)的模型
基于图神经网络的分子模型:
SchNet: arXiv:1706.08566
PhysNet: arXiv:1902.08408


如前所述,深度分子模型的实现,需要在框架层面进行多方面的适配,而在非常适合科学计算场景的MindSpore社区成立协调多个SIG协作的工作组,成为了深圳湾实验室分子动力学团队进行相关开源协作的首选。
WG-MM的主要职责将会是集中梳理分子动力学研究对MindSpore的需求、在每个版本周期同相关SIG合作落地相应诉求、以及形成相关的教程性文档。我们在此特别感谢深圳湾实验室团队选择MindSpore社区进行这样卓有远见的研究和开发。

1. 参加社区开发非常简单:
请先查看WG-MM的立项文档:
https://gitee.com/mindspore/community/blob/master/design/meps/mep-mm/MEP-MM.md
-
-
相关阅读:
Study--Oracle-05-Oracler体系结构
【【VDMA彩条显示实验之四 含C语言代码】】
【C++】布隆过滤器
ElasticSearch 实现分词全文检索 - SpringBoot 完整实现 Demo 附源码【完结篇】
TensorFlow的GPU使用相关设置整理
kubernetes code-generator使用
[附源码]Python计算机毕业设计SSM京津冀区域产学研项目管理信息系统(程序+LW)
Linux基础
【高级RAG技巧】在大模型知识库问答中增强文档分割与表格提取
[杂谈]-2023年实现M2M的技术有哪些?
- 原文地址:https://blog.csdn.net/Kenji_Shinji/article/details/126705765
- 最新文章
-
沪漂五周年了:我越来越迷茫了
Agentic Skill Routing 实战:别再把所有 Skill 塞进 AI Agent 上下文
MySQL-Seconds_behind_master的精度误差
[MAF预定义ChatClient中间件-03]CachingChatClient——利用缓存省钱省时间
AI的至暗历史:从万众期待到被政府撤资,AI的两次死亡徘徊
Agent OS :五种驯服不确定性的范式
PortSwigger SQL注入LAB11
数据库即时编译JIT
[Begin]AI Learn Data Day 0
深度学习进阶(二十七)现代 LLM 的核心架构设计其二:SwiGLU