-
AI为药物开发助力两篇文章介绍
Artificial Intelligence for drug discovery.
AI为药物开发助力
第一位演讲者:Dual-view molecule pre-training(双视图分子预训练)
paper:Search for Dual-view Molecule Pre-training | Papers With Code
主要贡献:
(1) To our best knowledge, we are the first to conduct molecule pre-training taking the advantages of the two different views (i.e., SMILES and molecular graphs).
(2) In addition to MLM, DMP leverages dual-view consistency loss for pre-training, which explicitly exploits the consistency of representations between two views of molecules.
Figure 1: Examples of molecular property prediction from MoleculeNet [55]. (a) Transformer succeeds while GNN fails; (b) GNN succeeds while Transformer fails; (c) both the standard Transformer and GNN fail while our method succeeds.
3.2 Training objective functions

Figure 2: The illustration of our dual-view consistency loss. "SG" indicates the stop-gradient when back-propagating. Our method consists of three training objective functions, including two masked language modeling (MLM) loss and one dual-view consistency loss.
(1) MLM on Transformer: Given a SMILES sequence, we randomly mask some tokens, and the objective is to recover the original tokens, following the practice in NLP pre-training [9, 35].
(2) MLM on GNN: Similar to the MLM on Transformer, we randomly mask some atoms in a molecular graph (the bonds remain unchanged), and the objective is to recover the original atoms, following the practice in graph pre-training [23].
(3) Dual-view consistency: To model the interaction between the GNN and the Transformer, we propose a dual-view consistency loss, that models the similarity of the output between GNN and Transformer. Based on the empirical discovery of [14], we introduce two non-linear projection layers
 ,
,  and two prediction layers
and two prediction layers 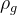 and
and 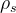 . For the SMILES view
. For the SMILES view 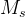 and the graph view
and the graph view of a molecule, we randomly mask some tokens/atoms and obtain
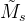 and
and 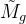 .
. 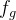 is obtained by pool
is obtained by pool 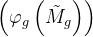 and fs is the first output of
and fs is the first output of 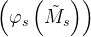 , which corresponds to the [CLS] token. After that, we apply the projection and prediction layers to them, i.e.,
, which corresponds to the [CLS] token. After that, we apply the projection and prediction layers to them, i.e.,
Since
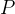 ·’s and
·’s and 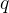 ·’s are the representation of the same molecule but from different views, they should preserve enough similarity. Let
·’s are the representation of the same molecule but from different views, they should preserve enough similarity. Let 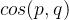 denote the cosine similarity between and , i.e.,
denote the cosine similarity between and , i.e., 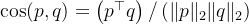 . Following [14], the dual-view consistency loss is defined as:
. Following [14], the dual-view consistency loss is defined as:
In Eqn.(2),
 means that the gradient is not applied to
means that the gradient is not applied to 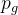 when back-propagating, and neither is
when back-propagating, and neither is 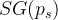 .
.After pre-training, we can use the Transformer branch, the GNN branch or both for downstream tasks. According to our empirical study, for molecular prediction tasks, we recommend using the Transformer branch. Using both branches brings further improvement at the cost of larger model size.
3.3 Discussions
(1) Relation with co-training: Considering there are two “views” in our method, people might be curious the relation with co-training, which also leveraged two views. Co-training [1] is a semi-supervised learning method that can be used to construct additional labeled data. The basic assumption to use co-training is that the features can be divided into two conditionally independent sets (i.e., views), where each set is sufficient to train a classifier. Each classifier provides the most confident predictions on the unlabeled data as the additional labeled data. In comparison, DMP is an algorithm which does not need the independent assumption. DMP provides a pre-trained model for downstream tasks instead of newly labeled data.(协同训练是半监督学习的方法为了生成新的标签数据,而DMP 是为下游任务提供预训练模型而不是新的标签数据)
In addition, there are some work leveraging multiple views for pre-training, but the views are usually from the same model instead of two heterogeneous ones (Transformer vs GNN). In [37], the two views are the outputs of a node-central encoder and an edge-central encode. In [17], the two views of a graph are the first-order neighbors and a graph diffusion. Those methods are significantly different from ours.
(2) Limitations of our method: Our method has two limitations. First, there is a Transformer branch and a GNN branch in our method, which increases the training cost comparing with previous single-branch pre-training [3, 53]. How to design an efficient pre-training method is an interesting future direction. Second, in downstream tasks, we deal with all molecules using either the Transformer branch or the GNN branch. Recent studies show that a better solution is to use a meta-controller to dynamically determine which branch to use [59, 12] for an individual input. We will also explore this dynamic branch selection in future.(在下游任务中,如何使用元控制动态决定用transformer还是GNN分支?)

为什么药物开发如此重要?因为它具有巨大的商业价值,预计到2025年底,全球药物发现市场预计到2025年将达到710亿美元。
但是,从发现一种新药到该药最终获得FDA批准,是一个非常漫长的过程。
- 如图所示,设计一种新药需要六年半的时间,并应用一些临床前实验(临床前实验是指某些细胞和动物实验不允许在人体上进行)。
- 在那之后,大约需要七年的时间进行临床试验,这意味着我们可以在人体上测试一种新药,但数量有限。
- 之后,FDA 将批准一种最成功的药物。
我们可以看到药物发现既昂贵又耗时,我们希望使用人工智能来帮助加快这一过程。

我们专注于第一部分,药物开发。在临床前之前有三件事。
①、首先是target discovery and validation。
在这里,target(靶点) 是我们体内的一些蛋白质或酶或核酸,它的活性可以被一些药物改变,从而产生特定的效果。在 target discovery中,给定一种特定的疾病,我们想尽最大努力找出所有可能的target ,这就是target discovery所做的。之后,我们使用target validation来过滤掉错误的target 。②、二是screening。
Virtual screening意味着我们使用计算机科学程序来计算药物与靶标的结合亲和力。我们搜索一些分子图书馆(molecular library),我们想找出一些可能的分子,以便将来可以将它们制成药物。这部分的主要技术包括对接和分子动力学(docking and molecular dynamics)。③、之后,我们进入lead generation and optimization process(生成和优化过程)。
这是因为在上一步中我们获得了一些分子,但效力仍然可以提高。我们想改进上一步的分子,在这个阶段,我们需要一些技术,包括生成新分子(generating new molecules),我们需要一些关于分子性质预测(property prediction)的技术,我们需要一些关于逆合成( retrosynthesis)的技术。分子建模
我们可以看到,在上述三个阶段中,分子建模(molecule modeling)是上述所有步骤的重要任务。所以我们想朝这个方向努力,我们提出了一种新技术,针对这些任务的双视图分子预训练(dual-view molecule pre-training)。
在进入技术细节之前,我们应该知道如何表示一个分子:

我们可以将 2D 图形转换为 1D SMILES 字符串,这是分子的基本表示。
考虑到一个分子至少有两种表示,相应地,就有两种模型:

1、分子建模的第一种方式(通过1D的 SMILES mask)
如果我们把一个分子看作一个 SMILES 字符串,我们可以使用transformer来处理这个分子。让我们在这里以蛋白质为例。为了输入它的 SMILES 序列,我们随机屏蔽了一些标记,并使用transformer重建屏蔽标记。
2、分子建模的第二种方式(通过2D的图像mask)
我们也可以将分子视为二维图。我们还可以mask一些原子和字母 G 模型来恢复被屏蔽的原子。
Dual-view molecule pre-training(双视图分子预训练)

基本上,有两种类型的技术。它们很好,但以前,这两种模型是独立使用的。但这就是一切吗?让我们看看一些案例。在这里我们进行了丰富的实验,并在这里展示了一些典型的例子。在左侧部分,在分子性质预测任务中,GNN 在左侧的两个分子上成功,但transformer失败。对于右侧的两部分,transformer 成功,但 GNN 失败。
我们可以说 GNN 实际上偏爱结构丰富的分子。例如,三个以上的环串联在一起,但不利于链较长的分子。相比之下,transformer 偏爱相对较长的分子,但不偏爱具有重组的分子。
它们是互补的,受此启发,我们提出了一个新模型,将这两种模型结合在一起。这就是我们称为双视图分子预训练的解决方案。

- 在我们的框架中,一个输入分子由两个视图表示,一个是sequence视图,它是一个SMILE字符串,一个是graph视图,它是一个 2D 图形表示。
- 我们将SMILE输入到Transformer中,并将graph输入到GNN中。
- 我们首先将math language modeling(MLM)损失应用于Transformer和 GNN 模块。
- Transformer 分子和 GNN 模块可以输出同一个小分子的特征,因为这些特征来自同一个小分子,它们在某些latent space中不应该相似。受此启发,我们使用余弦相似度(cosine similarity)来衡量它们在某些latent space中的相似度,并迫使两个模型最小化两种表示的差距。

之后,我们在一些任务上尝试我们的模型。第一个任务称为molecular property prediction,数据集是molecular net,是分子性质预测的benchmark dataset。
我们对七项任务进行了实验,我们在这里只展示了三个结果。
- 这三个任务是BBBP,这是一个关于预测血脑屏障、药物浓度的任务,
- 第二个任务是ClinTox,它与预测药物的毒性有关。
- 第三项任务是预测抑制HIV 复制的能力。
我们可以看到,与之前的baseline相比,与仅使用质transformer相比,与仅使用 GNN 相比,我们的方法在所有这些方法中取得了最好的效果。这显示了我们方法的有效性。

让我们看看一些案例(case)。表现怎么样?
使用我们提出的模型后,我们可以看到该模型可以处理具有长链和相对重组(long chains and relatively restructures)的分子。这表明了我们提出的方法的有效性。

除了预测任务,我们还将我们的模型应用于逆合成(retrosynthesis)(任务是给定一个目标分子,我们想用一些相对容易获得的小分子来制造目标)。
使用我们提出的方法后,我们可以看到在两个基准数据集上,我们的方法进一步改进了基线并在这两个任务上取得了最先进的结果。这再次显示了我们方法的有效性。

对于未来,有很多有趣的方向,
- 首先,我们将在药物开发方面尝试multitasks,
- 其次是我们将设计更先进的模型。例如,如何设计更强大的 GNN 模型,以及如何使用单个模型同时涵盖 Transformer和 GNN。
- 第三,我们将尝试更多的数据。目前,我们使用了1 亿个数据,我们希望在未来尝试更多。
第二位演讲者:Accurate drug-target inaction(DTI) prediction for drug discovery
我将介绍另一个关于药物发现的研究项目,Accurate drug-target inaction(DTI) prediction for drug discovery. (药物开发的准确药物靶点不作为预测)。

疾病与人类之间的战争从未结束。传统的药物发现过程从最初的药物发现到获得FDA的批准是非常漫长的,通常需要10年以上的时间。因此,使用计算机辅助药物开发(也称为CADD)可以节省大量时间和金钱消耗。CADD 可以分为四个步骤。
首先是目标的识别和验证(targeted identification and validation);
第二个是筛选(screening);
第三个我们需要优化候选药物的性质和设计成功的姿势。所有候选药物都应通过正确的实验和临床试验进行严格评价。虽然 CADD 可以降低药物发现的成本,但准确性和生成性不能满足我们的要求,因为不同的targets和drug之间变化很大。
在我们的研究中,我们专注于第二步,药物的虚拟筛选(Drug virtual screening)和设计
我们的算法是为了提高准确性和泛化能力。

如右上图所示Drug virtual screening是从庞大的数据集中选择active候选药物,其中包含数十亿种化合物。这个任务可以分为两个步骤:
首先是主动结合预测(active binding prediction),从数百万化合物选择可以与蛋白质靶标结合的候选药物。
其次,对于主动位置预测(active pose prediction),我们需要确定它们的native并施加到相应的靶蛋白上。
如右下图所示,受体蛋白以cartoon表示,药物以stake表示。Ligand配对体(Drug)与Receptor受体(Protein)结合的区域称为结合袋(binding pocket)。
我们的目标是预测药物是否可以与蛋白质结合,如果可以,则确定binding poses.。对于这项任务,关键挑战是如何表示和区分以下三种交互:
- Intramolecular information of receptor(受体分子内信息)
- lntramolecular information of ligand(配对体分子内信息)
- lntermolecular information between ligand and receptor(配体与受体之间的分子间信息)

最经典的方法是Molecular docking(分子对接):
- 如右图所示,docking是一种分子动力学(molecular dynamics);
- 估计配体是否能与靶受体结合;
- 预测靶点结合袋内配体的结合构造

随着深度学习的发展,提出了许多新的算法,可以分为两类。
第一个是将分子内和分子间相互作用作为一个整体:
- Take the whole structure of drug-target docked pose as input(以药物-靶标docked pose的整体结构为输入)
- Both for active binding and active pose tasks()
- GatedGAT (Lim,2020) uses graph attention and achieves the best performance(其中之一是 GatedGAT,它使用图注意力并获得更好的性能)
- Limitation: cannot distinguish intermolecular interact from intramolecular interactions explicitly(然而,这种方法不能明确区分分子间相互作用和分子内相互作用)
另一种方法分别模拟配体和受体的分子间相互作用,而MONN是这种方法的最佳算法。
- Do not use the docked pose(没有使用docked pose,因此无法显式建模分子间信息)
- Trim the ligand and receptor respectively
- MOON (Li, 2021) uses several convolutional modules and achieved the best performance
- Limitation: only for active binding task; do not explicitly use the intermolecular information from docket pose
但是,由于它不使用停靠姿势,因此无法显式建模分子间信息。
因此,在我们的研究中,我们提出了Intermolecular graph transformer,它使用three-way深度神经网络分别对三种相互作用进行建模。

我们的 IGT 模型可以分为三个部分。
- 首先是一个feature extraction module (特征提取模块),直接从新设计的 atom(原子) 和 bond features 中获取分子间信息;
- 其次是 message passing module(消息传递模块),具有重复构建块的 three-way module(三向模块),分别模拟配体内的相互作用、受体内的相互作用以及配体和受体之间的相互作用;
- 最后是readout module(读出模块预测最终输出)。


值得注意的是,由于第二种方法不使用docked put 作为输入,因此无法生成active pose。在对active pose prediction进行评估时,与 GatedGAT 和对接相比,IGT在所有指标上都取得了最佳性能。鉴于 IGT 的性能,我们进一步使用它来识别针对 SARS-CoV-2 主要蛋白酶的活性结合药物。

- COVID19 是由病毒 SARS-CoV-2 引起的;
- 进行Virtually screening 以识别针对SARS-CoV-2主要蛋白酶的活性结合药物,所有数据均经过wet实验验证;
- IGT 模型成功检测到针对 SARS-CoV-2 的near-native pose;
- 预测的结合pose形成类似的相互作用,也可以在原始复杂结构中找到。
如左图所示,作为ground truth的预测绑定pose分别为绿色和蓝色。受体蛋白的结合口袋被Surface 染成粉红色。我们预测的绑定姿势非常接近原生结构。因此,IGT模态成功地检测了针对 SARS-CoV-2 的接近原生姿势。此外,在绑定姿势之前的中间和红色面板形成了类似的相互作用,这些相互作用也可以在原生结构中找到。

鉴于多色工业在医疗保健中的重大科学意义和巨大的应用潜力,我们的方法有望为工业客户提供药物发现调查。其次,海量的化合物数据具有巨大的计算密集型任务,既要提供巨大的存储空间,又要提供巨大的计算资源,因此,此类技术的错误可用性也将为微软提供来自微软云的小商机。最后,我们的协议预测了具有适当结合姿势的活性结合药物,用于健康解决方案药物防御,它们有助于加工药物。
https://www.microsoft.com/en-us/research/video/research-talk-ai-for-drug-discovery/
-
相关阅读:
go-fastdfs安装(国产分布式文件系统)
高级深入--day31
计算机网络-传输层(UDP协议报文格式,伪首部,UDP校验过程)
20和遍历以及迭代器有关的一些东西
【C++】泛型编程
极坐标和直角坐标的雅克比矩阵推导
虹科工业树莓派应用案例之在石油开采中的应用
【ChatGPT系列】ChatGPT:创新工具还是失业威胁?
换掉ES!Redis官方搜索引擎来了,性能炸裂!
LeetCode笔记:Biweekly Contest 87
- 原文地址:https://blog.csdn.net/weixin_43135178/article/details/126458906