-
喜报!易基因“同源基因特异性甲基化时序数据分析方法”获专利授权
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
喜报!深圳市易基因科技有限公司发明的《一种同源基因特异性甲基化时序数据的分析方法》获专利授权。该方法首先利用DNA甲基化测序数据序列特征和突变信息判定ASM区域,筛选出每个时期或者组别中特有的所述ASM区域,之后结合不同突变型对应的甲基化信息,利用某个时期的甲基化信息和其他不同时期样品进行比较,初步判定出原始时期特异性ASM区域,最后通过统计所有所述原始时期特异性ASM区域内的差异以及全部CpG位点数目,采用超几何检验,判定所述ASM区域是否为最终的时期特异性同源基因特异性甲基化区域。本发明进一步深化分析挖掘甲基化信息,可以检测多个不同阶段基于重亚硫酸盐处理的数据的ASM动态变化。
专利背景:
在DNA甲基化数据分析的过程中,特别是针对WGBS(全基因组甲基化)数据,很多人都觉得无从下手,常规的分析思路通常是找DMR(Differentially methylated regions,差异性甲基化区域),并进行注释分析;但是此方案挖掘出来的信息非常有限,大量相关的DNA甲基化信息被遗漏。
ASM(Allele-Specific DNA Methylation,等位基因特异性甲基化),除了我们通常关注的等位基因特异性甲基化与印记基因和女性X染色体的关系外,研究表明等位基因特异性甲基化影响等位基因间甲基化差异高达85%的完整CpG岛。等位基因间的甲基化差异与表型强烈相关。等位基因特异性甲基化能够导致甲基化的基因拷贝的等位基因抑制。同时,等位基因特异性甲基化可能影响了约10%的全部人类基因,并促进了等位基因特异性表达和单等位基因沉默。因此,等位基因特异性甲基化代表了遗传多态性如何能够导致表型变异的一条表观遗传途径。研究表明等位基因特异性甲基化是一种表观遗传漂变的结果,其方向由等位基因间的异常差异确定,同时获得一个等位基因高甲基化的倾向被遗传。
现有ASM确定的方法有两个类别,首先是通过针对BS数据进行SNP分型,选择突变频率接近50%的位置确定其不同突变型的DNA甲基化率差异,另外一种思路是通过DNA片段设计二项式分布模型,并结合贝叶斯方法判定ASM区间,要求区域内片段读长的甲基化分布表现为明显的两个类型的甲基化模式,并且两种模式的数量基本一致,如amrfinder软件,但是此种方法只针对单个样品;现阶段并没有适合判定多个阶段或者多个分组之间比较ASM特征以及ASM变化过程的方法。
深圳市易基因科技有限公司开发的《一种同源基因特异性甲基化时序数据的分析方法》突破了相关技术的瓶颈。该分析方法是易基因科技自2015年开始研发的技术,于2018年申请发明专利,并于2022年获得发明授权。

专利申请只是技术的原型和起点,几年来易基因持续对方法进行优化和升级。
专利技术实例
本实施例利用9例猪全基因组甲基化数据对方法进行测试,利用该方法判定的结果与实验预期的结果一致。
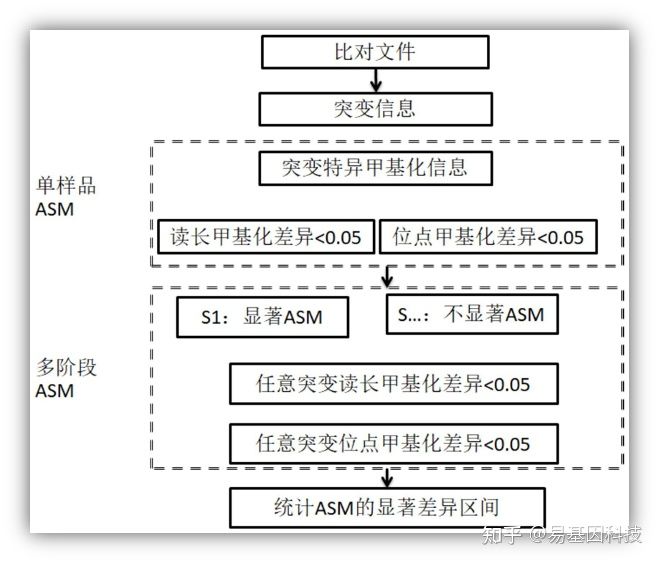
图1:实例分析方法流程图
(1)将所检测的9例经过重亚硫酸盐测序数据与猪参考基因组比对、排序,去除冗余测序数据;此实验数据中9例猪全基因组甲基化数据共分为前中后三个时期,每个时期三个重复;
(2)利用CpG位点的重亚硫酸盐转化C的个数信息得到CpG位点上覆盖到的C的个数R(m)以及经过重亚硫酸盐转化后的T的个数信息R(t);以及甲基化率Methy_Rate=R(m)/(R(m)+R(t));
(3)利用甲基化数据得到每个样品的核苷酸突变信息,利用相应的突变信息将有关reads分为两类(A,B),且每类的比例在0.3~0.7之间;分别统计区间内每条reads的对应位置的CpG位点的甲基化状态,定义读长中测序为C的位点为甲基化C(m),测序为T的位点为非甲基化C(n),并进一步统计测序读长的甲基化水平,methy_reads(i)=C(m)/(C(m)+C(n)),i∈A U B;同时判定所述两类reads中的甲基化率在CpG位点上的甲基化情况methy_rate
(Ai),methy_rate(Bi);利用t检验判定所述两类reads的甲基化率差异,若同时满足p(methy_reads(i))<0.05、p(methy_rate(i)) <0.05,为保证统计有效性,该过程筛选CpG为点数>5;满足上述筛选条件则判定该区域为等位基因特异性甲基化区域(ASM);此处为保证实验数据的可靠性,结合本申请的研究目的为前中后三个时期特异性的ASM的变化,所以此处将三个时期的样品ASM结果取交集;其交叠情况如图2‑4所示;从图2‑4可以看出,三个时期中的ASM区间在重复样品中大量重合,重合部分即为排除样品特异性时期相关ASM数据集。

图2:样品前期三个重复之间的交叠情况

图3:样品中期三个重复之间的交叠情况

图4:样品的后期的三个重复之间的交叠情况
(4)利用步骤(3)中筛选出的所述方法在研究中多个时期的组别分别进行统计,筛选出多个时期中只有单独一个时期特有的ASM区域为待选时期特异性ASM区间U(ASM i ),并筛选出所有位点的CpG位点的甲基化信息;进一步利用核苷酸突变信息分别统计各个时期的测序读长的甲基化信息reads_stage(I,A or B),对应位点的甲基化信息Methy_rate_stage(I,A or B),分别统计时期特异性的差异情况methy_reads(I,A or B),对比reads_stage(I,A or B);methy_rate(I,A or B),对比Methy_rate_stage(I,A or B);分别采用t检验p(reads_stage,A or B)<0.05、p(Methy_rate_stage,A or B) <0.05的结果确认疑似阶段特异性ASM区间U(ASMi)。
(5)筛选步骤(4)中CpG甲基化信息,统计每个特异性ASM区间中的CpG位点的甲基化率在每个样品中的甲基化率,分别比较A、B两种突变类型至少一种对应的reads的甲基化率有差异的位点个数k(ASMi),以及区间内的CpG个数m(CGi),总数为N,以及所有的疑似阶段特异性ASM区间U(ASMi)中的总数n:

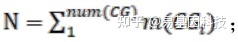
(6)采用超几何分布判别相应的区域内的富集情况,取p<0.05的ASM区域为时期特异性ASM区间:

并对相应的区间做基因注释,最终取前中后三个时期独有的基因集用于后续研究,在此实施案例中,我们发现前期特异性ASM对应1539个特异性基因,中期对应1076个,后期对应基因3111个,结果见图5。

图5:ASM对应基因区域在前中后三个时期的情况图。大量的基因区域有重合,但在每个时期都有每个时期独自的ASM区域存在,说明在青春期发育过程中等位基因特异性甲基化的发生是一个动态的过程。
(7)对步骤(6)中的前期、中期、后期三个时期的相关基因进行GO和KEGG注释,确定所述相关基因中ASM对应的基因在整个发展过程中或者组间比较的过程中的功能层面的变化。
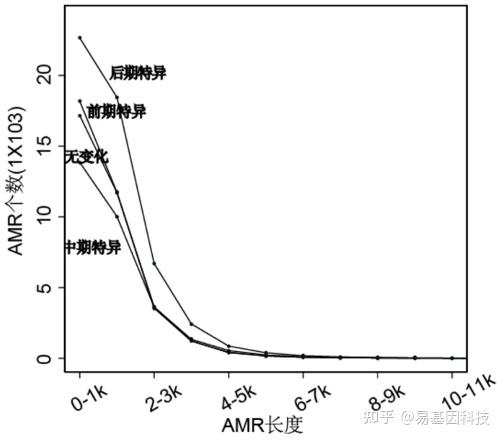
图6:合并后ASM区间长度与个数的分布情况图。发生等位基因特异性甲基化的区域集中在3k以内。
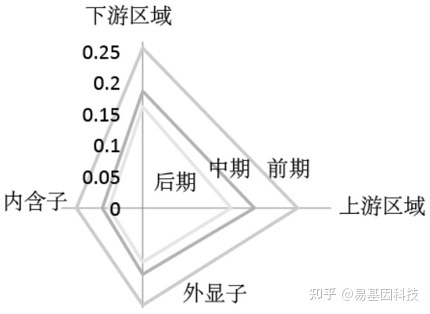
图7:ASM区域在基因上的位置分布情况图。针对各个时期特异性的ASM进行分析,从ASM区域在各个基因元件上的分布可以看出,每个时期发生在各个基因元件上的区域分布情况也有明显的改变。

图8:样品在三个时期特异性ASM的甲基化水平分布图。通过进一步考察这些区域的DNA甲基化,发现对应的甲基化率相较与其他时期都比较低,这也从反面印证了检验区间的正确性。
-
相关阅读:
查看linux系统情况常用命令
Vue中this.$set()解决页面不更新问题
ADC测试杂谈一:配置基于matlab+quartus的测试环境
桌面应用开发:Go 语言和 Web 技术的融合创新 | 开源日报 No.46
PyTorch搭建CNN-LSTM混合模型实现多变量多步长时间序列预测(负荷预测)
Java项目:JSP会议-会议室管理系统
CSS-06:多类名的使用场景
【成像光敏描记图提取和处理】成像-光电容积描记-提取-脉搏率-估计(Matlab代码实现)
分享两个实用的PPT制作技巧
测试开发(6)软件测试教程——自动化测试selenium(自动化测试介绍、如何实施、Selenium介绍 、Selenium相关的API)
- 原文地址:https://blog.csdn.net/E_gene/article/details/126384563
 https://link.zhihu.com/?target=https%3A//mp.weixin.qq.com/s%3F__biz%3DMzAwNTY3NDIxMw%3D%3D%26mid%3D2650651534%26idx%3D1%26sn%3D79b6caa8cfbb79dd50e6f7c32f0278ae%26chksm%3D83101e04b467971228eba9657f9bb7a97cafaecbc5e52d4f4cefe961e015fad3a0f7a6b802fc%26token%3D1639287666%26lang%3Dzh_CN%23rd
https://link.zhihu.com/?target=https%3A//mp.weixin.qq.com/s%3F__biz%3DMzAwNTY3NDIxMw%3D%3D%26mid%3D2650651534%26idx%3D1%26sn%3D79b6caa8cfbb79dd50e6f7c32f0278ae%26chksm%3D83101e04b467971228eba9657f9bb7a97cafaecbc5e52d4f4cefe961e015fad3a0f7a6b802fc%26token%3D1639287666%26lang%3Dzh_CN%23rd