-
如何进行DAP-seq的数据挖掘,筛选验证位点
从样本准备到寄送公司,每一天都在“祈祷”有个心仪的分析结果,终于在这天随着邮件提示音的响起,收到了分析结果......
分析前工作
爱基在进行数据分析之前,会有两次质控报告反馈给老师们。第一个,基因组DNA的提取质控报告(图1):保证DNA的完整性以及足够的量进行后续的富集亲和纯化;第二个,富集建库报告:构建DNA文库,利用磁珠富集与加完halo Tag标签表达的目的蛋白结合DNA片段,并纯化获得IP文库。这个过程中,为了检测蛋白表达的正常,爱基利用抗体对富集产物进行 WB 检测,同样对于文库也会进行质检(图2)。


图1 DNA提取质控报告

图2 WB结果显示目的蛋白表达正常
分析思路
第一部分
数据预处理:去接头序列、污染序列、低质量碱基,获得clean data序列,并进行相关数据统计;
第二部分
参考基因组比对:将clean data定位到参考基因组上,得到bam文件,并去除重复序列,保留唯一比对的序列;
第三部分
call peak: 将bam文件进行Peak检测,得到富集区域的信息,并进行Peak在基因功能元件的分布,最近基因寻找及motif预测。
第四部分
Peak分析:统计Peak分布情况,对Peak最近基因进行GO、KEGG功能注释与富集及转录因子预测等。
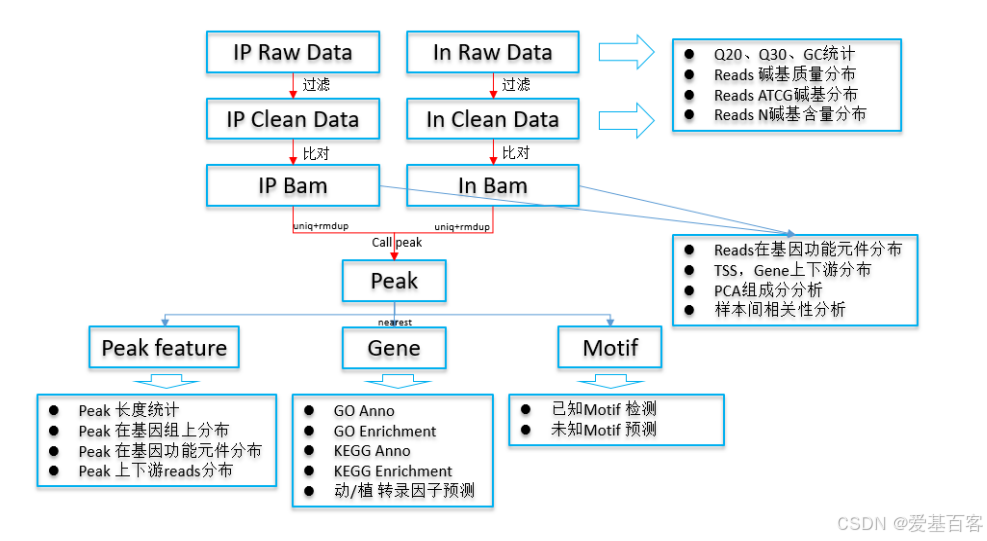
图3 DAP分析流程
纵览整个本地分析结果,peak和motif可谓是重中之重。爱基结果“03.peak”中包含了peak的长度统计、peak在功能元件分布饼图、peak在基因组上的分布情况(是否有染色体偏好)以及关键peak的reads分布图,以上这些分析图也是在文献中普遍会见到的。而“06.motif”的结果则包含了大量潜在结合基序信息,从中老师们可以筛选到心仪的验证位点。
如何筛选验证位点
1. 从基因角度出发
在“03.peak/01.peak_annotation”表格中记录着peak的详细信息,包括:在染色体上具体位置、长度、峰顶所在染色体的位置、显著性、富集倍数、落在某个基因的哪个位置、统计距离最近基因以及这些基因的在不同数据库的注释结果。
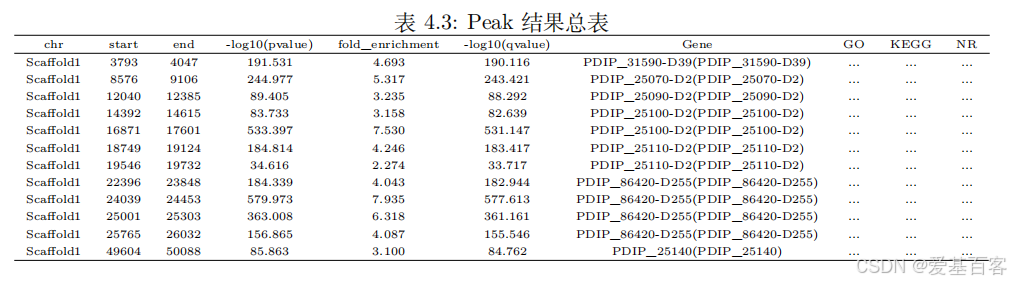
如果前期做过其它实验或者通过文献查找已经有了关注基因,那么直接搜索基因id找到对应的peak,通过获得的peak编号在“06.Motif”文件夹的ecxel表格中找到匹配Peak的motif就可以考虑验证啦~
如果没有做过上述调查,可以现在基因注释列(GO、KEGG、NR......)搜索与自己课题相关的关键词。比如,抗旱研究可以搜索活性氧、激素(ABA、GA)等。锁定到与研究内容相关的gene,同行对应上peak,再和上述方式一致根据peak找到motif。
总之,这种方式逻辑是从gene→peak→motif。
2. 直接锁定基序
可以直接看motif网页版结果中的match Details,有无基序在数据库中已经被收录匹配目标转录因子(homerResults中看Best Match/Details;KnownResults中看Name列)。
以“sna/MA0086.2/Jaspar(0.681)”为例,其含义是这个比对结果来自Jaspar数据库的sna转录因子,MA0086.2是Jaspar的编号,可通过这个具体编号找到对应sna-motif信息(当没有MA编号时,可以直接搜索转录因子的名称),0.681代表该denovo motif与这个sna-motif的序列相似打分。如果研究的是sna就可以优先关注这个基序啦。
除此之外,软件会自动按照显著性排序,将更显著的排在前列;碱基复杂程度低的、只有2个碱基不断重复的,不建议优先考虑哦。
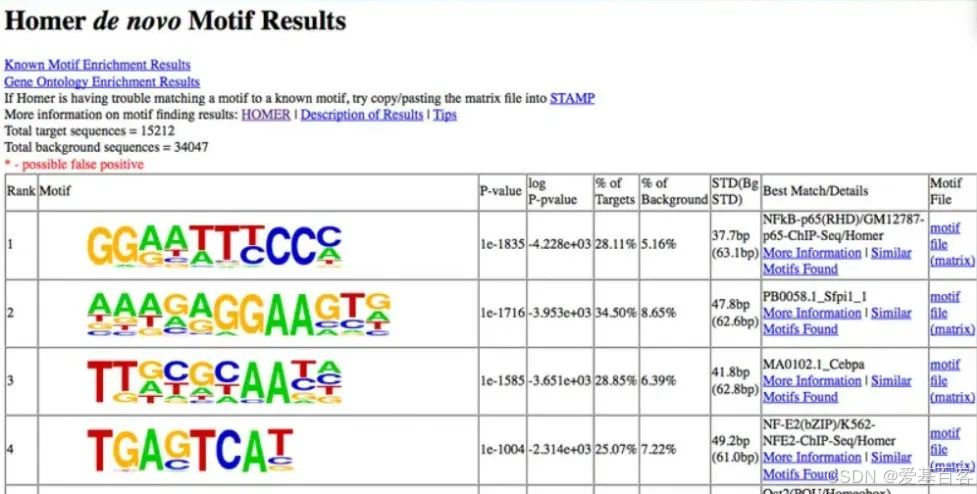
注:Known和homer 是两种不同的motif预测算法,结果都是可信的。Known motif基于已有转录因子数据库的motif结果,比对本次的peak有没有在这些已有的研究motif上富集;homer result是指利用所有的peak从头(de novo)计算得到motif,然后会比对已有转录因子数据库的motif,看比对率最一致的是哪个(bestmatch)。两者不一定一致(因为motif序列是一组序列模式,相似的序列可能会被归为同一个motif)。
扩 展
通过上述的方式已经锁定了想要验证的基因位点后,还需要确定下motif在基因/基因启动子区真实存在的碱基信息哦。参考:【干货分享 | 一文GET寻找motif在序列上的定位】
想要更多了解,欢迎各位老师前来咨询哦~

-
相关阅读:
Aveva Marine VBNET 编程系列-搭建开发框架
Ajax同源和跨域和节流防抖
科技云报道:分布式存储红海中,看天翼云HBlock如何突围?
部署lamp
数组模拟环形队列(思路分析) [数据结构与算法][Java]
sql server 查询某个字段是否有值 返回bool类型
数学建模学习(82):模拟退火算法(SA,matlab版本)
活动报名与缴费小程序开发笔记一
Android学习笔记
大数据请把这个分离软件推给所有后期~
- 原文地址:https://blog.csdn.net/Igenebook/article/details/142135937