-
GSEA -- 学习记录
brief
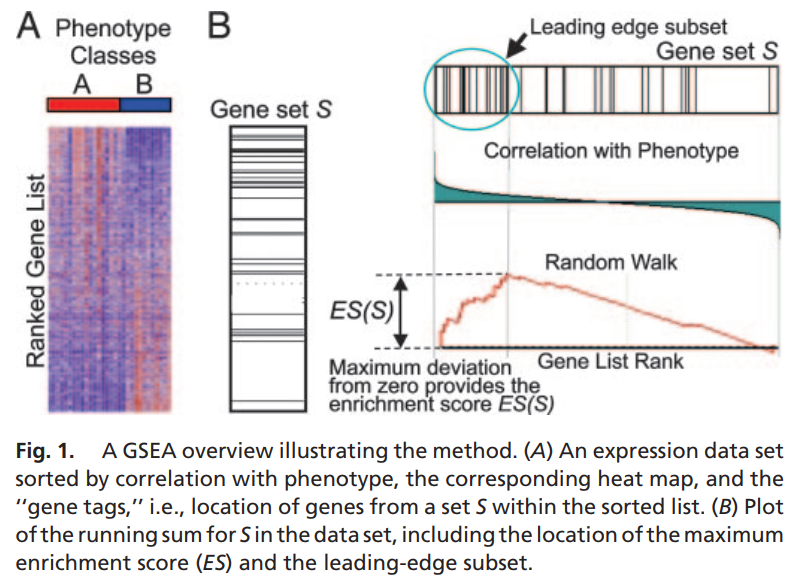
上一篇学习记录写了ORA,其中ORA方法只关心差异表达基因而不关心其上调、下调的方向,也许同一条通路里既有显著高表达的基因,也有显著低表达的基因,因此最后得到的通路结果对表型的解释力度也不大。还有一些基因表达量的变化程度很小,但是其生物学功能可能很重要,那么该如何体现?
GSEA方法让通路的上下调分析成为可能,简单来说,先计算基因在不同组间表达量的log2FC,并以此从大到小进行排序,这样就得到了一个基因从上调到不变到下调的基因列表。然后,对于我们感兴趣的基因集,我们只需考察它们的成员是否富集在这个基因列表的上方(上调部分)或者下方(下调部分)即可判断这些基因集的上下调情况。
or like this:

优点:1.相比起GSA,GSEA 可以实现不再仅关注于差异基因,可以不受p-value以及log2FC阈值的影响,可以获得更多生物学功能变化的信息。
2.富集分数ES,实际上是k-s like test的统计量,所以ES主要表示基因集S的基因的log2FC的分布与不在基因集S的其他基因的log2FC的分布是否一致,当ES大于0并且具有统计学意义时,那我们可以说基因集S内基因相比其他基因表达上调。统计学原理部分
需要补充的基础知识:
https://blog.csdn.net/jiangshandaiyou/article/details/136545010其他注意事项
- GSEA中为了强调表型的变化,也就是表型间基因表达量的log2FC,算法调整了eCDF每个阶梯的上升高度。
在标准k-s的eCDF中,阶梯上升高度是1/n,而在GSEA中,考虑到log2FC的权重,基因集S的eCDF的上升高
度随着S内不同基因的log2FC变化:
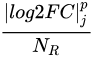

因此,在eCDF中,基因集S中的基因越是有着较大|log2FC|,其上升的阶梯就越大。
- 上面描述了在基因集S中的基因的上升阶梯高度,而不在基因集S中的其他基因的上升阶梯并不受log2FC的权重影响,并且与标准k-s检验的eCDF一致:

N表示所有基因的数量,NH表示感兴趣的基因集(基因集S)中包含的基因个数。所以,简单表示其上升阶梯高度即为:

- 随着x轴(ranked gene list)的位置不断变化,enrichment scores也在发生变化。ES的数学表达方式为,对基因集S的每个gene的eCDF求和。当ES为正时表示基因集S的基因富集在ranked gene list 的顶部,ES为负时表示基因集S的基因富集在ranked gene list 的底部。
转录组部分
library(clusterProfiler) library(org.Hs.eg.db) library(AnnotationDbi) # step1 # get ranked gene list df <- read.table("../out.tpm.csv",header = T,sep = ",",row.names = 1) names <- rownames(df)[order(df$PBMC,decreasing = T)] DEG_ranked <- sort(df$PBMC,decreasing = T) names(DEG_ranked) <- names head(DEG_ranked) # MT-CO1 MT-ND4 MT-CO3 MT-CO2 MT-ND1 MT-ND2 # 35393.65 23674.00 22698.25 18457.70 17377.82 15141.74 # 所以最重要的输入ranked gene list 是一个整数型的向量,而且以gene symbol 作为 name # step2 # get KEGG pathway informations library(msigdbr) hs_msigdb_df <- msigdbr(species = "Homo sapiens") # Filter the human data frame to the KEGG pathways that are included in the curated gene sets hs_kegg_df <- hs_msigdb_df %>% dplyr::filter( gs_cat == "C2", # This is to filter only to the C2 curated gene sets gs_subcat == "CP:KEGG" # This is because we only want KEGG pathways ) # step3 # Run gsva gsea_results <- GSEA( geneList = DEG_ranked, # Ordered ranked gene list minGSSize = 10, # Minimum gene set size maxGSSize = 500, # Maximum gene set set pvalueCutoff = 0.05, # p-value cutoff eps = 0, # Boundary for calculating the p value pAdjustMethod = "BH", # Benjamini-Hochberg correction TERM2GENE = dplyr::select( hs_msigdb_df, gs_name, gene_symbol ) ) head(gsea_results@result) # gsea_result_df <- data.frame(gsea_results@result) # Visualizing results most_positive_nes_plot <- enrichplot::gseaplot( gsea_results, geneSetID = "HALLMARK_MYC_TARGETS_V2", title = "HALLMARK_MYC_TARGETS_V2", color.line = "#0d76ff" ) most_positive_nes_plot- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- 44
- 45
- 46
- 47
- 48
- 49
- 50
- 51
- 52
- 53
- 54
- 55
- 56
- 57
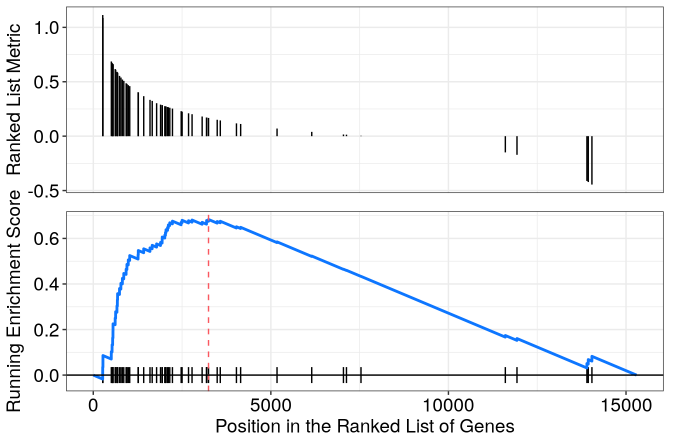
单细胞部分
#BiocManager::install("clusterProfiler",force = TRUE) library(clusterProfiler) library(Seurat) # 这里面有一个小的数据集 pbmc_small ,一会儿用它试手 library(tidyverse) library(reshape2) # getwd() setwd("D:/") data("pbmc_small") Idents(pbmc_small) Idents(pbmc_small) <- pbmc_small@meta.data$groups deg <- FindMarkers(pbmc_small,ident.1 = "g1",ident.2 = "g2",min.pct = 0.01, logfc.threshold = 0.01,thresh.use = 0.99, assay="RNA") # 排序:根据log2FC rank高表达和地表达的基因 geneList <- deg$avg_log2FC names(geneList) <- toupper(rownames(deg)) geneList <- sort(geneList,decreasing = T) head(geneList) # 获取gene set # gmt 文件在MsigDB下载的 geneset <- read.gmt("h.all.v2023.2.Hs.symbols.gmt") # head(geneset) length(unique(geneset$term)) egmt <- GSEA(geneList, TERM2GENE=geneset, minGSSize = 1, pvalueCutoff = 0.99, verbose=FALSE) # head(egmt) egmt@result gsea_results_df <- egmt@result rownames(gsea_results_df) write.csv(gsea_results_df,file = 'gsea_results_df.csv') # BiocManager::install("enrichplot",force = TRUE) <- 这里可视化 library(enrichplot) gseaplot2(egmt,geneSetID = 'HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION',pvalue_table=T) gseaplot2(egmt,geneSetID = 'HALLMARK_MTORC1_SIGNALING',pvalue_table=T)- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
- 14
- 15
- 16
- 17
- 18
- 19
- 20
- 21
- 22
- 23
- 24
- 25
- 26
- 27
- 28
- 29
- 30
- 31
- 32
- 33
- 34
- 35
- 36
- 37
- 38
- 39
- 40
- 41
- 42
- 43
- GSEA中为了强调表型的变化,也就是表型间基因表达量的log2FC,算法调整了eCDF每个阶梯的上升高度。
-
相关阅读:
mpvue小程序 vant组件单选
Kubernetes-常用命令
AWS SAA-C03 #207
Redis(11)Hyperloglog
MySQL 索引类型和存储引擎详解
uniapp x — 跨平台应用开发的强大助力
Spring实例化源码解析之FactoryBean(十一)
【工具篇】高级 TypeScript 案例
获取网卡上的IP、网关及DNS信息,获取最佳路由,遍历路由表中的条目(附源码)
【ArcGIS微课1000例】0054:尺寸注记的创建与编辑
- 原文地址:https://blog.csdn.net/jiangshandaiyou/article/details/134457515
