-
GEO生信数据挖掘(五)提取临床信息构建分组,分组数据可视化(绘制层次聚类图,绘制PCA图)
检索到目标数据集后,开始数据挖掘,本文以阿尔兹海默症数据集GSE1297为例
上节做了很多的基因数据清洗(离群值处理、低表达基因、归一化、log2处理)操作,本节介绍构建临床分组信息。
我们已经学习了提取表达矩阵的临床信息
- # 安装并加载GEOquery包
- library(GEOquery)
- # 指定GEO数据集的ID
- gse_id <- "GSE1297"
- # 使用getGEO函数获取数据集的基础信息
- gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = FALSE ,getGPL = F)
- #提取临床信息 方法一:$或者@ ,配合str()观察结构
- pdata = gse_info$GSE1297_series_matrix.txt.gz@phenoData@data
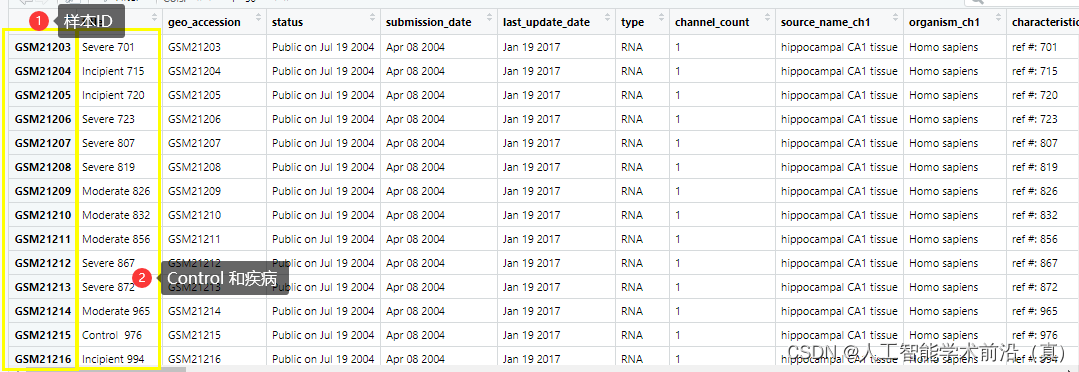
临床信息预处理
提取出关键的两列
- #构建样本分组信息
- group_data = pdata[,c('geo_accession','title')]
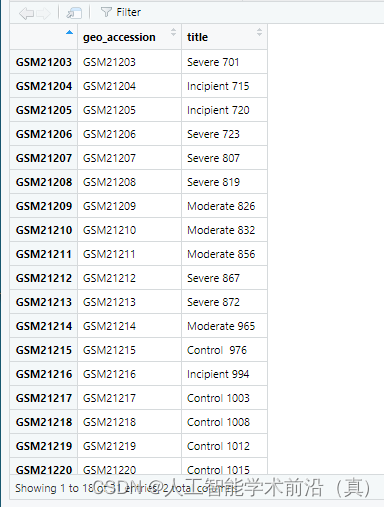
本例中的疾病和对比组的标识比较复杂,要考虑剔除数字,还要考虑做2分类还是4分类。
字符串处理二分类
- # 使用grepl函数判断字符串是否包含'abc',并进行相应的修改
- group_data$group_easy <- ifelse(grepl("Control", group_data$title), "Control", "AD")
字符串处理四分类
- # 使用grepl函数判断字符串是否包含特定内容,然后进行相应的修改
- group_data$group_more <- ifelse(grepl("Control", group_data$title), "Control",
- ifelse(grepl("Moderate", group_data$title), "Moderate",
- ifelse(grepl("Incipient", group_data$title), "Incipient","Severe")))
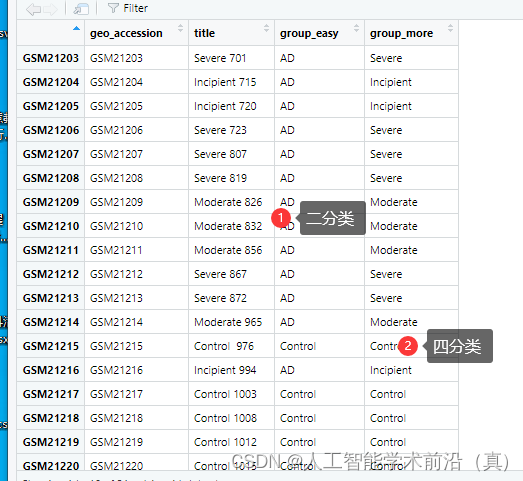
处理后的结果,无需调整分组信息的顺序,让AD在一堆,Control在一堆,现在的<临床信息表的行索引GSM顺序>与<基因表达信息表的列索引GSM顺序> 是一致的。
需要的分组信息已经提取完毕。
分组后箱线图可视化
上一节保存了数据清洗后的基因表达矩阵,加载进来,为了使用简单boxplot画图,我们又增加了一列区分不同样本类型的颜色。
- #加载基因表达矩阵
- load("exprSet_clean_75percent_filter.RData") #exprSet_clean
- # 使用grepl函数判断字符串是否包含'Control',并进行颜色标记,为画图
- group_data$group_color <- ifelse(grepl("Control", group_data$title), "yellow", "blue")
- #(3)画箱线图查看数据分布
- group_list_color = group_data$group_color
- boxplot( data.frame(exprSet_clean),outline=FALSE,notch=T,col=group_list_color,las=2)

分组后层次聚类图可视化
- exprSet =exprSet_clean
- #修改GSM的名字,改为分组信息
- colnames(exprSet)=paste(group_data$group_easy,1:ncol(exprSet),sep = '')
- #定义nodePar
- nodePar=list(lab.cex=0.6,pch=c(NA,19),cex=0.7,col='blue')
- #聚类
- hc=hclust(dist(t(exprSet))) #t()的意思是转置
- #绘图
- plot(as.dendrogram(hc),nodePar = nodePar,horiz = TRUE)

分两类好像看不太出来,聚类的好坏,我们又观察了分四类后的聚类情况,效果不错。

聚类的效果还不错,没有特别别扭的分类。
分组后PCA图可视化
由于样本量比较少,看起来,两类样本,在空间上还算分的比较开。(后期可以把轻微症状放到Control组,做测试看看效果。)
至此为止,临床信息预处理工作完毕,基因表达数据预处理工作完毕,最让人头疼的工作结束。
最后别忘了保存一下根据临床数据构建的分组信息,后面的差异分析要用哦
-
相关阅读:
分布式架构的高性能与可用性
Robot Framework 自动化测试详解
Spring通过配置文件管理Bean对象
jstl标签传参数失败
Spring Boot 内置工具类介绍
【阿里云】ssl证书到期更新
Linux_应用篇(08) 信号-基础
新手如何免费创建一个自己的网站?
如何从宏观层面构建优秀的大语言模型
netty群聊客户端服务器及心跳检测
- 原文地址:https://blog.csdn.net/zzh1464501547/article/details/133364899