-
qPCRtools | 神仙R包分分钟搞定你的qPCR实验结果!~
1写在前面
不知道大家都是怎么完成
qPCR的计算的,在不会R的时候,我是用一个祖传的Excel表进行计算的。🤣
但是,一直有个缺点,如果需要计算的量比较大时,就不方便了,去搜了一下文献,发现了一个最近发表的R包,不仅可以计算反转录的RNA体积,还可以帮助选择定量方法,简直是神仙R包,本期就介绍一下它的使用吧。🥰
感谢原作者的开发,嘿嘿,文末有引用方法。👀2用到的包
rm(list = ls())
library(tidyverse)
library(ggsci)
library(qPCRtools)
library(ggstatsplot)- 1
3计算反转录用的RNA体积
3.1 示例数据
包内自带了示例数据,这里我们就直接加载吧。🥳
df.1需要至少2列,sample和concentration,剩下的大家随意。🤣
Note! 这里浓度默认是ng/ul。🤜df.1.path <- system.file("examples", "crtv.data.txt", package = "qPCRtools")
df.1 <- data.table::fread(df.1.path)
head(df.1)- 1

Note! 这里我们的
df.2文件至少要包含一个all的列,告诉R具体的反应体积。🤒df.2.path <- system.file("examples", "crtv.template.txt", package = "qPCRtools")
df.2 <- data.table::fread(df.2.path)
head(df.2)- 1

3.2 开始计算
现在我们就知道每个
sample该如何配置反转路体积啦,Perfect!😁
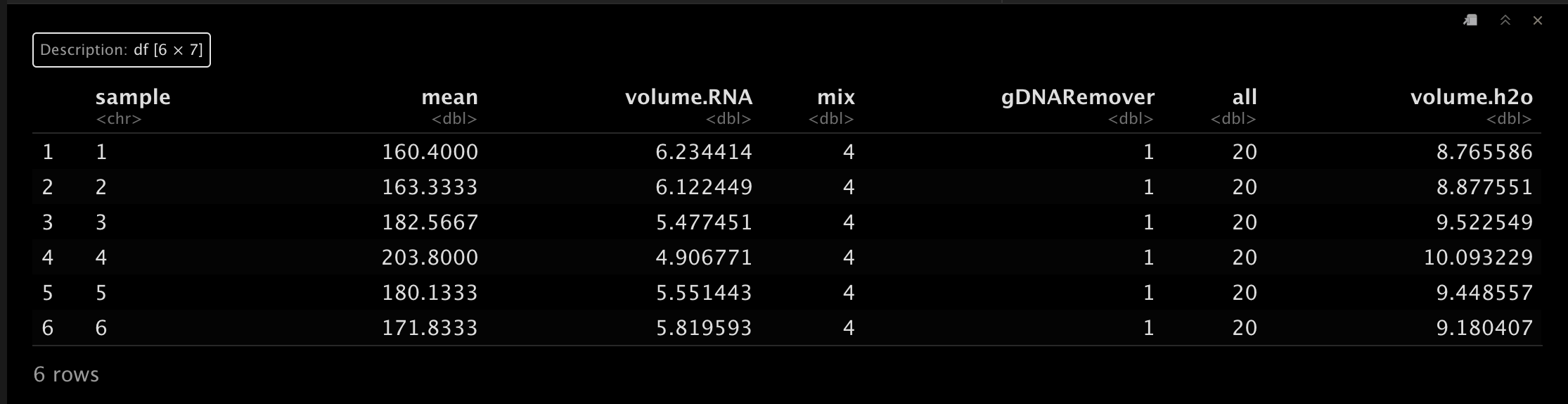
这里我们假设反转1ug。🤩result <- CalRTable(data = df.1, template = df.2, RNA.weight = 1)
head(result)- 1
4相对标准曲线和扩增效率的计算
拿到新的
Primers应该先进行扩增效率的计算,一起看下怎么弄吧。👇4.1 示例数据
df.1包含至少2列,孔的位置和Cq值。😗df.1.path <- system.file("examples", "calsc.cq.txt", package = "qPCRtools")
df.1 <- data.table::fread(df.1.path)
head(df.1)- 1

df.2包含至少2列,孔的位置和浓度。🫠df.2.path <- system.file("examples", "calsc.info.txt", package = "qPCRtools")
df.2 <- data.table::fread(df.2.path)
head(df.2)- 1

4.2 开始计算
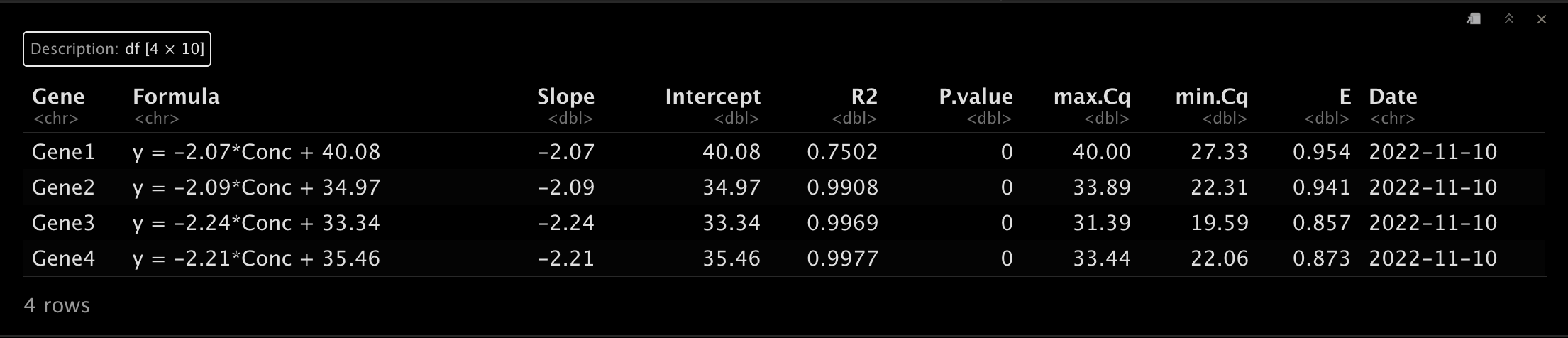
Note! 大家注意一下这里的稀释倍数,默认是
4,可以按需更改。😂CalCurve(
cq.table = df.1,
concen.table = df.2,
lowest.concen = 4,
highest.concen = 4096,
dilu = 4,
by = "mean"
) -> p
p[["table"]]- 1
4.3 可视化
p[["figure"]] +
theme_bw()+
scale_color_npg()- 1

5使用相对标准曲线法计算基因表达水平
如果内参基因和目的基因的
扩增效率不相等,我们就不能使用2-ΔΔCt法了,需要选择无参的方法。🤨5.1 示例数据
cq.table至少包含position和Cq值。😘df1.path <- system.file("examples", "cal.exp.curve.cq.txt", package = "qPCRtools")
cq.table <- data.table::fread(df1.path)
head(cq.table)- 1

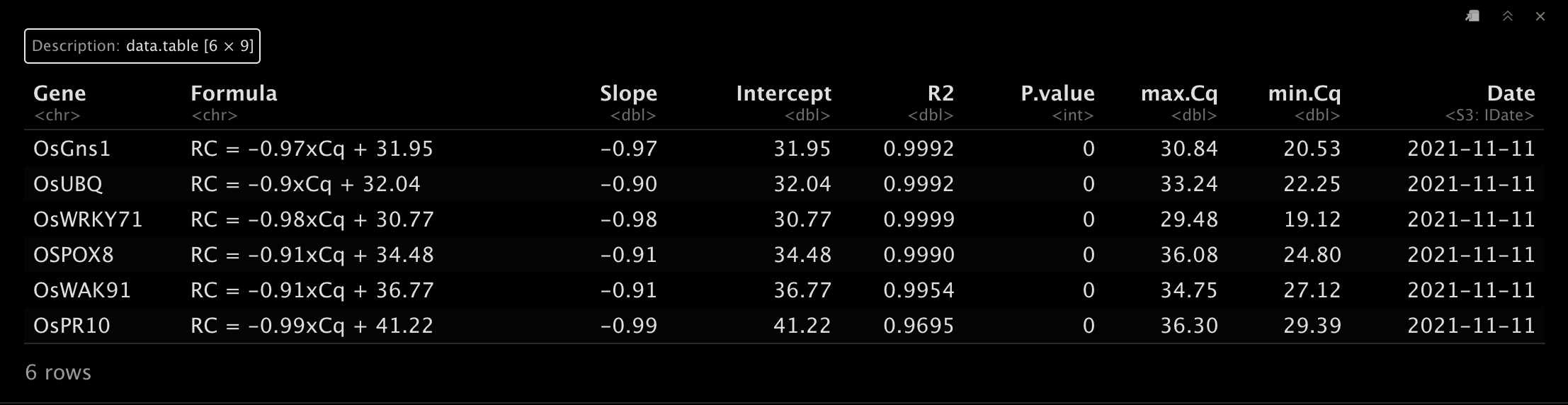
curve.table为标准曲线,可以通过前面介绍的方法计算得出。😂df2.path = system.file("examples", "cal.expre.curve.sdc.txt", package = "qPCRtools")
curve.table = data.table::fread(df2.path)
head(curve.table)- 1
design.table需要包含position和相应的信息,如干预、基因名等。 🙃df3.path = system.file("examples", "cal.exp.curve.design.txt", package = "qPCRtools")
design.table = data.table::fread(df3.path)
head(design.table)- 1

5.2 开始计算
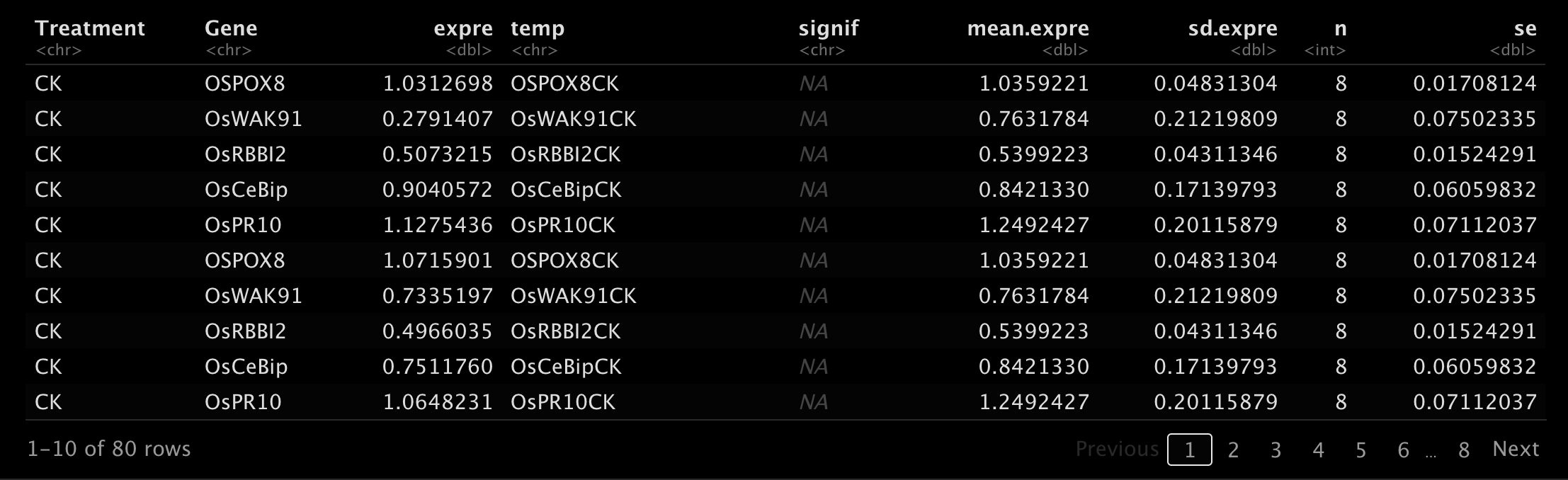
CalExpCurve(
cq.table,
curve.table,
design.table,
correction = TRUE,
ref.gene = "OsUBQ",
stat.method = "t.test",
ref.group = "CK",
fig.type = "box",
fig.ncol = NULL) -> res
res[["table"]]- 1
5.3 可视化
大家可以直接使用
res[["figure"]]提取结果的可视化图,这里我为了更加美观,提取了数据进行美化。😘res[["table"]] %>%
grouped_ggbetweenstats(y = expre,
x = Treatment,
grouping.var = Gene,
type = "nonparametric"
)- 1

62-ΔΔCt法计算表达水平
数据准备与上面的方法相似,这里就不做具体介绍了。😂
6.1 示例数据
df1.path <- system.file("examples", "ddct.cq.txt", package = "qPCRtools")
cq.table <- data.table::fread(df1.path)
head(cq.table)- 1

df2.path <- system.file("examples", "ddct.design.txt", package = "qPCRtools")
design.table <- data.table::fread(df2.path)
head(df.2)- 1
6.2 开始计算
CalExp2ddCt(cq.table,
design.table,
ref.gene = "OsUBQ", ## 内参
ref.group = "CK", ## 对照
stat.method = "t.test", ## 统计方法
fig.type = "bar",
fig.ncol = NULL) -> res
res[["table"]]- 1

6.3 可视化
res[["table"]] %>%
grouped_ggbetweenstats(y = expre,
x = Treatment,
grouping.var = gene,
type = "nonparametric"
)- 1

7使用RqPCR方法计算表达水平
这种方法也是一种不需要内参的计算方法,数据格式也是几乎一样的。
7.1 示例数据
df1.path <- system.file("examples", "cal.expre.rqpcr.cq.txt", package = "qPCRtools")
cq.table <- data.table::fread(df1.path, header = TRUE)
head(cq.table)- 1

df2.path <- system.file("examples", "cal.expre.rqpcr.design.txt", package = "qPCRtools")
design.table <- data.table::fread(df2.path, header = TRUE)
head(design.table)- 1
7.2 开始计算
CalExpRqPCR(cq.table,
design.table,
ref.gene = NULL,
ref.group = "CK",
stat.method = "t.test",
fig.type = "box",
fig.ncol = NULL
) -> res
res[["table"]]- 1
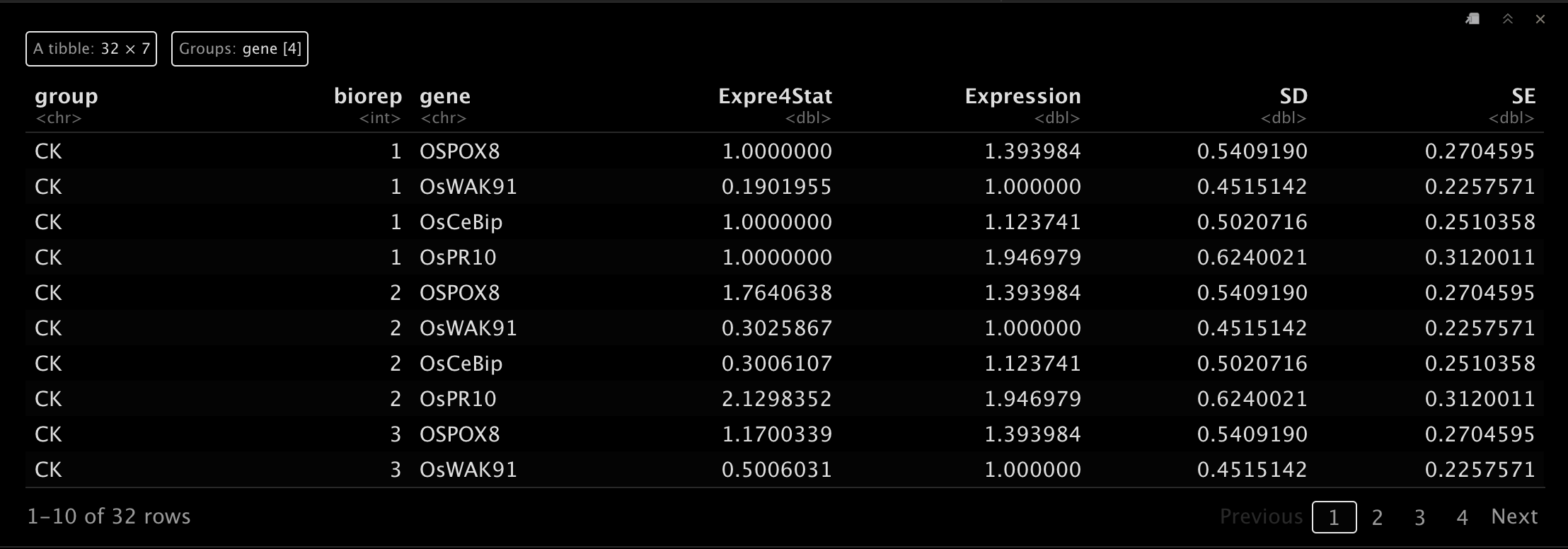
7.3 可视化
res[["table"]] %>%
grouped_ggbetweenstats(y = Expre4Stat,
x = group,
grouping.var = gene,
type = "nonparametric"
)- 1

8引用
🌟 如何引用:👇
Li X, Wang Y, Li J, Mei X, Liu Y, Huang H. qPCRtools: An R package for qPCR data processing and visualization. Front Genet. 2022;13:1002704. Published 2022 Sep 13. doi:10.3389/fgene.2022.1002704

最后祝大家早日不卷!~
点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 往期精彩 📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......本文由 mdnice 多平台发布
-
相关阅读:
nginx端口映射后,跳转带的是内网端口而不是内网端口
项目管理软件dhtmlxGantt配置教程(三):配置树列
202.快乐数
MySQL日志管理和权限管理(重点)
课堂问题:一个凸函数的性质
Unity之ShaderGraph如何实现水波纹效果
Python中RotatingFileHandler、TimedRotatingFileHandler函数用法
如何评价GPT-4o?
40个高质量SSM毕设项目分享【源码+论文】(三)
D. Cyclic Operations Codeforces Round 897 (Div. 2)
- 原文地址:https://blog.csdn.net/m0_72224305/article/details/127793749