-
主成分分析PCA并给出解释百分比
出图:包括PC1和PC2的散点图,以及PC1和PC2的解释百分比。
1. 处理思路
思路:
1,根据plink文件,进行pca分析
2,根据特征值,计算pca1和pca2的解释百分比
3,根据特征向量结果,进行pca作图2. 注意事项
注意:
特征值就是特征向量在对应维度的方差,特征值所占所有特征值之和的比值,就是其对应特征向量的方差贡献率。
简单来说:
- PCA1是特征向量,其方差是PC1的特征值,其方差贡献率为PC1特征值的百分比
- PCA2是特征向量,其方差是PC2的特征值,其方差贡献率为PC2特征值的百分比
3. 示例演示
示例:
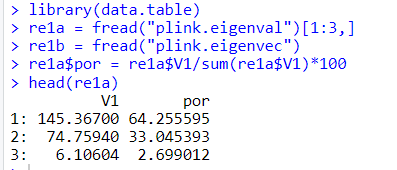
比如计算一个plink进行文件的3个pca,结果如下:plink --bfile geno/b --pca 3- 1
- 2
结果包括:
- plink.eigenval ,特征值,共有3行数据,分别是3个PCA的特征值
- plink.eigenvec,特征向量,第三四五列是3个PCA的特征向量,作图用前两个PCA
$ head plink.eigenvec 0 ID1 -0.032 0.0185407 0.0351135 0 ID2 -0.0330665 0.0213082 0.0575101 0 ID3 -0.0340043 0.0209365 -0.00264537 0 ID4 -0.0323621 0.0203962 0.0503156 0 ID5 -0.0325016 0.0191183 0.0426273 0 ID6 -0.0346765 0.0196053 -0.0408817- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
$ head plink.eigenval 145.367 74.7594 6.10604- 1
- 2
- 3
- 4
- 5
4. 计算PCA百分比
如果想要十分精确的计算每个PCA的得分,那我们需要计算所有PCA的值,PCA的个数等于样本的个数。
比如我们的样本有575个,那么它计算PCA的代码为:
plink --bfile geno/b --pca 575- 1
- 2
可以看到,样本数和pca的行数都是575行
$ wc -l geno/b.fam 575 geno/b.fam $ wc -l plink.eigenvec 575 plink.eigenvec $ wc -l plink.eigenval 575 plink.eigenval- 1
- 2
- 3
- 4
- 5
- 6
- 7
在R语言中计算每个PCA的百分比,以及PCA可视化:
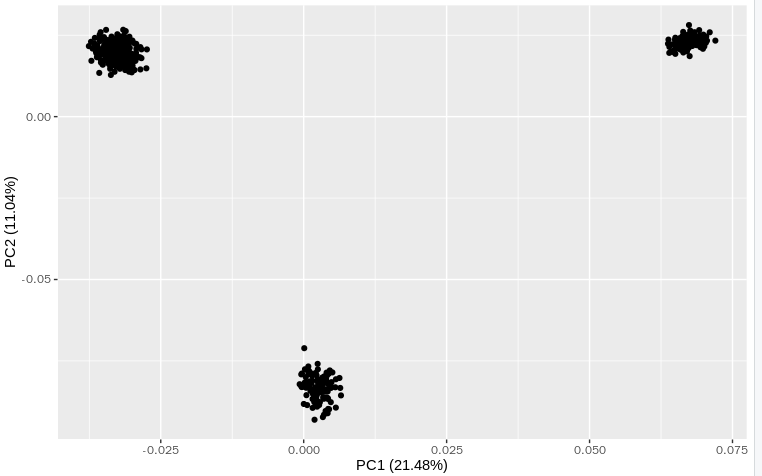
library(tidyverse) library(tidyverse) re1a = fread("plink.eigenval") re1b = fread("plink.eigenvec") re1a$por = re1a$V1/sum(re1a$V1)*100 head(re1a) ggplot(re1b,aes(x = V3,y = V4)) + geom_point() + xlab(paste0("PC1 (",round(re1a$por[1],2),"%)")) + ylab(paste0("PC2 (",round(re1a$por[2],2),"%)"))- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
结果:
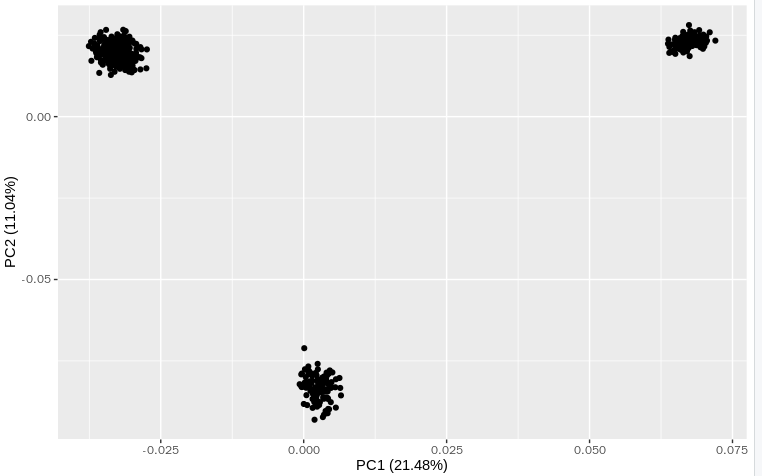
5. 使用前10个做PCA百分比计算
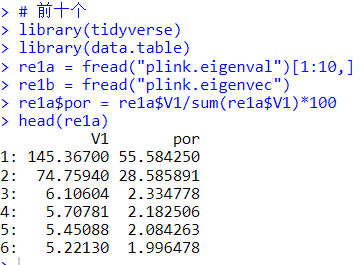
因为PCA的特征向量从大到小排列,所以,也可以用前3个或者前10个作为代表,计算PC1和PC2的百分比,我们测试一下:
取前三个
这个偏差太大了,PC1从原来的21%,到现在的64%,不靠谱!
取前十个
也不靠谱,变化也比较大,还是老老实实的用所有的特征值去计算百分比吧,麻雀虽小,积土成山呀!
取所有
这个才是最正确的!
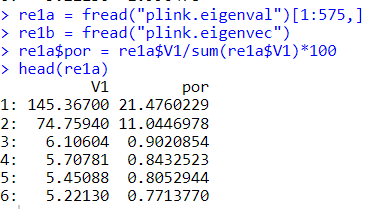
6. 一步到位
现在的问题是,样本的个数,还要查看,然后定义–pca number,再读取,可以在R中一步到位:
思路:
- 读取plink文件的fam,确定个数
- R中调用plink,传参个数
- 作图
args="geno/b" nn = dim(fread(paste0(args[1],".fam"),header=F))[1] system(sprintf("~/bin/plink --bfile %s --allow-extra-chr --chr-set 30 --pca %s",args[1],nn)) re1a = fread("plink.eigenval") re1b = fread("plink.eigenvec") re1a$por = re1a$V1/sum(re1a$V1)*100 head(re1a) ggplot(re1b,aes(x = V3,y = V4)) + geom_point() + xlab(paste0("PC1 (",round(re1a$por[1],2),"%)")) + ylab(paste0("PC2 (",round(re1a$por[2],2),"%)"))- 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
- 11
- 12
- 13
-
相关阅读:
数位DP 上 day44
【开发技术】2万字详细介绍Docker 和 web项目的部署监控,docker部署,拉取kafana,prometheus镜像监控
kotlin的suspend对比csharp的async&await
TCP/IP(七)TCP的连接管理(四)全连接
stm32,STC89C51使用串口下载程序
基于LVM通过添加硬盘实现分区扩容的方法介绍
机器学习_10、集成学习-随机森林
Redis集群操作-----主从互换
【go】go 实现行专列 将集合进行转列
Spark - Task 与 Partition 一一对应与参数详解
- 原文地址:https://blog.csdn.net/yijiaobani/article/details/127770834