-
TCGA里面的任意基因做生存分析 批量生存分析
欢迎关注微信: 生信小博士
在刚刚进入生信领域的时候,我想做的事情就是三个,
第一,知道任何我想研究的基因在组织中的表达情况,
第二,我选的基因对肿瘤的生存有无影响,
第三,这个基因可能的作用是什么?
这是来自临床医生的视角,研究疾病,最终希望能够服务临床,临床离不开诊断和治疗,假设一个基因的表达对肿瘤的预后有影响,他很可能就是我的盘中餐。
有一大堆网页工具可以实现生存分析,但是你看看jimmy已经写的帖子
一拳把人打翻在地,扶都扶不起来。 但是没有办法他是群主。即使会随时被朝阳群众扭送到派出所,他依然是群主,他的排版有问题,但是架不住他的内容有深度,群主喜闻乐见。
那么问题来了,上面的问题也可以反过来问,既然别人已经准备好了网页工具给你用,你还写代码做什么?!
四个字,批量,自由
大气一点就是高端定制。
就这么简单。 那开始我们的表演:
今天我们要对TCGA里面的任意基因做生存分析,最关键的我们要批量做生存分析,然后选取生存差异最显著的基因。
首先安装需要的包:
-
# Load the bioconductor installer. -
source("https://bioconductor.org/biocLite.R") -
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/") -
# Install the main RTCGA package -
biocLite("RTCGA") -
# Install the clinical and mRNA gene expression data packages -
biocLite("RTCGA.clinical") -
biocLite("RTCGA.mRNA")
然后是加载包
-
library(RTCGA) -
#了解数据 -
infoTCGA <- infoTCGA() #这个命令会返回一个数据框,可以知道有哪些数据可被下载 -
#获得临床数据: -
# Create the clinical data -
library(RTCGA.clinical) -
clin <- survivalTCGA(BRCA.clinical) #到这里临床部分的信息已经获得啦
得到数据后我们先看一下他是什么结构
-
class()
[1] "data.frame"
再看一下前面几行数据
-
head(clin)
times bcrpatient barcode patient.vital_status
1 3767 TCGA-3C-AAAU 0
2 3801 TCGA-3C-AALI 0
3 1228 TCGA-3C-AALJ 0
4 1217 TCGA-3C-AALK 0
5 158 TCGA-4H-AAAK 0
6 1477 TCGA-5L-AAT0 0
简单说就是三列,TCAGid,生存时间,和发生的事件
获得gene表达数据:
-
library(RTCGA.mRNA) #加载数据包 -
class(BRCA.mRNA) #查看数据类型发现是个数据框 -
dim(BRCA.mRNA) #看一下数据维度发现有590个样本,17815个基因 -
BRCA.mRNA[1:5, 1:5] #看一下部分数据样子
bcr_patient_barcode ELMO2 CREB3L1 RPS11 PNMA11 TCGA-A1-A0SD-01A-11R-A115-07 0.5070833 1.43450 0.765000 0.52600
2 TCGA-A1-A0SE-01A-11R-A084-07 0.1814167 0.89075 0.716000 0.13175
3 TCGA-A1-A0SH-01A-11R-A084-07 0.4615000 2.25925 0.417125 0.32500
4 TCGA-A1-A0SJ-01A-11R-A084-07 0.8770000 0.43775 0.115000 0.75775
5 TCGA-A1-A0SK-01A-12R-A084-07 1.4123333 -0.63725 0.492875 0.94325
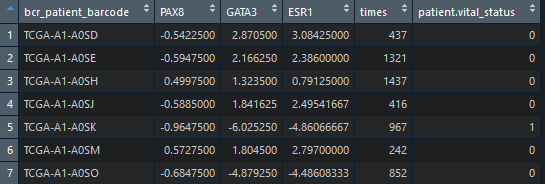
好了,行是样本,列为gene 下面我们挑选几个基因的表达数据出来,融合到生存的数据上去,因为表达量数据中TCGA的id号要长一点 所以在融合前,需要先裁剪一下,为了避免产生过多的中间变量,我们使用管道符号%>%,他的作用是 把前一个计算得到结果,作为第二个函数的参数,示例如下:
-
library(dplyr) -
exprSet <- BRCA.mRNA %>% -
# then make it a tibble (nice printing while debugging) -
as_tibble() %>% -
# then get just a few genes,这里是测试用 -
select(bcr_patient_barcode, PAX8, GATA3, ESR1) %>% -
# then trim the barcode (see head(clin), and substr) -
mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) %>% -
# then join back to clinical data -
inner_join(clin, by="bcr_patient_barcode")
看一下数据,发现就是表达量加上time,event两列,所以记住这规律,最终批量的时候需要减掉这两列
mark
开始做生存分析
-
library(survival) -
library(survminer)
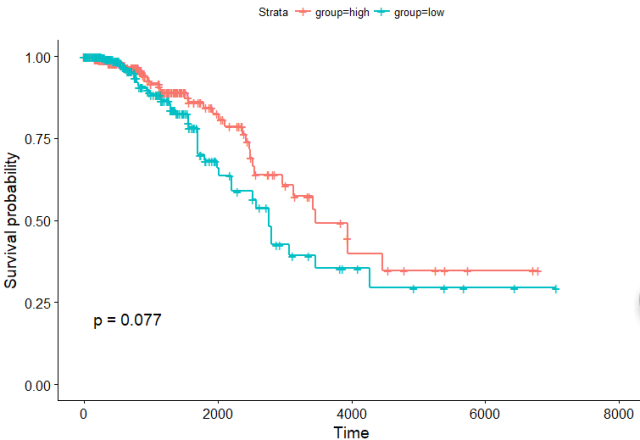
对需要做生存分析的样本分组,把连续变量变成分类变量,这里选择测试的基因是GATA3
-
group <- ifelse(esprSet$GATA3>median(esprSet$GATA3),'high','low')
构建生存对象,并且进行数据处理,这里是两步,我只是融合在了一起
-
sfit <- survfit(Surv(times, patient.vital_status)~group, data=esprSet) -
sfit -
summary(sfit)
直接绘图
-
ggsurvplot(sfit, conf.int=F, pval=TRUE)
mark
单个基因绘制成功,我看一下能不能小规模循环操作 首先得到得到生存对象my.surv
-
my.surv <- Surv(esprSet$times, esprSet$patient.vital_status)
使用apply循环,对数据的2到4列进行操作,实际上就是PAX8, GATA3, ESR1这三个基因, 这里面使用了survdiff,用来比较差异大小,获得p值
-
log_rank_p <- apply(esprSet[,2:4], 2, function(values1){ -
group=ifelse(values1>median(values1),'high','low') -
kmfit2 <- survfit(my.surv~group,data=esprSet) -
#plot(kmfit2) -
data.survdiff=survdiff(my.surv~group) -
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1) -
})
运行完了之后返回的找出小于0.05的P.val,在这个例子里面因为没有基因的p.val小于0.05,所以筛选不出来
-
log_rank_p <- log_rank_p[log_rank_p<0.05]
筛选后排序,并获得基因名 genediff <- as.data.frame(sort(logrank_p))
好了生存数据已经获取,
表达数据小规模试验可行,
批量操作也能实现,
下面就进入实战环节,前戏实在太长,但是你要相信你只要不睡着,这些都是值得的
获得完整的表达量数据
-
library(dplyr) -
esprSet <- BRCA.mRNA %>% -
# then make it a tibble (nice printing while debugging) -
as_tibble() %>% -
# then trim the barcode (see head(clin), and ?substr) -
mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) %>% -
# then join back to clinical data -
inner_join(clin, by="bcr_patient_barcode")
构建生存对象my.surv
-
library(survival) -
my.surv <- Surv(esprSet$times, esprSet$patient.vital_status)
在进行下一步之前,我居然突发奇想,我想看一看,这个表达数据里面哪些基因的NA值最多,有没有NA值多过样本数量一般的基因呢? 先构建了一个函数,他对数据的列起作用,统计NA值的个数,最终返回成一个数据框
-
rem <- function(x){ -
r <-as.numeric(apply(x,2,function(i) sum(is.na(i)))) -
return(data.frame(geneName=names(x)[which(r > 0)],na_num=r[which(r > 0)])) -
}
然后对表达量数据进行统计
-
na_count <- rem(esprSet)
最终发现NA最多的基因是LCE1B,有17个,所以数据不需要特殊处理啦
-
na_count <- dplyr::arrange(na_count,desc(na_num))
那就开始批量运算了,一开始就用apply,发现大概需要运行50分钟以上,所以尝试使用并行化处理 R语言里面的并行化有个专门的项目就是给apply的,使用起来也是很方便
-
#尝试使用并行运算 -
library(parallel) -
#detectCores()检查当前电脑可用核数 -
cl.cores <- detectCores() -
#makeCluster(cl.cores)使用刚才检测的核并行运算,我的服务器是28核56线程,我就用50吧 -
cl <- makeCluster(50) -
#这是坑,parApply里面用到的函数以及变量都需要申明,不声明就必须用模块 -
clusterExport(cl,c("esprSet","my.surv")) -
#length(names(esprSet))-2,为什么减去2,因为之前小规模测试时,我们知道最后两个是time和event,不是表达量 -
#数据从25开始,原因是从2开始会报错,暂时无法解决,还有要注意是parApply,A要大写的 -
log_rank_p <- parApply(cl,esprSet[,25:length(names(esprSet))-2],2,function(values){ -
group=ifelse(values>median(na.omit(values)),'high','low') -
kmfit2 <- survival::survfit(my.surv~group,data=esprSet) -
#plot(kmfit2) -
data.survdiff=survival::survdiff(my.surv~group) -
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1) -
})
这个运行的时间可能就是2分钟 终止并行化
-
stopCluster(cl)
找出小于0.05的P.val
-
log_rank_p <- log_rank_p[log_rank_p<0.05]
筛选后排序,并获得基因名
-
gene_diff <- as.data.frame(sort(log_rank_p))
最终得到的gene是2153个,这个数据是我之前留下的,本次写贴时要带孩子,在家没法运行运算。 数据保存可以这样:
-
save(gene_diff,file = "gene_df.Rda")
如果想用的时候就这样:
-
load(file = "gene_df.Rda")
没有必要先写成txt格式,然后要用的时候再读取进来,直接保存成R语言的格式即可,
你能想象每次用完word保存成图片,下次再使用时用OCR识别图片变成文字再编辑的状态么?
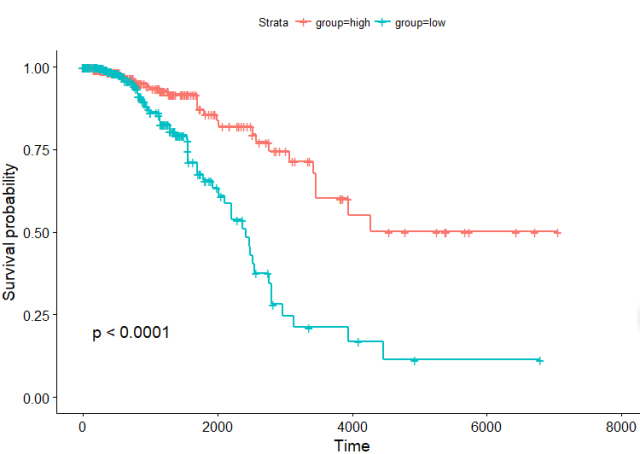
既然到了这一步,我们随便选取一个基因来作图,闭着眼睛都知道,都是有差异的
-
library(survminer) -
group <- ifelse(esprSet$LRRC8D>median(esprSet$LRRC8D),'high','low') -
sfit <- survfit(Surv(times, patient.vital_status)~group, data=esprSet) -
ggsurvplot(sfit, conf.int=FALSE, pval=TRUE)
mark
好吧效果很不错嘛
这时候把癌和癌旁的数据作差异分析,得到的基因与今天获得的基因取交集,就可以获得又差异表达,又对生存有影响的基因了。
到了这里,我用两个帖子,解决了我一直以来困扰我的两个问题。
也就是我只剩下一个问题了:
我找到的基因调控了谁,谁又来调控他呢?这个问题十分有难度,实现起来也很困难。
好在,我碰到了嘉因生物的小哈,小丫,她在他们的微信号里写了一系列这方面的帖子,我很受启发,但是技术方面要求很高,我在当时实现不了。
幸好!我又碰到生信技能树,跟着他们我开始学习RNAseq,ChIPSeq,ATAC-seq还有HiC,我要成为专业的生信工程师。
所以,今年有望彻底解决这个问题!
那么我的心愿也就了了,我搞这么久就是为了满足自己的心愿。
那时候我可以这么说,我是唯一一个能够同时解决这三个问题的临床医生。
那么,下面的问题来了,入门生信时有个特别烦人的拦路虎,就是软件安装。
-
-
相关阅读:
mybatis面试题及回答
java并发-锁
ubuntu 怎样查看隐藏文件
Mybatis-Plus复习
微信小程序中下载xlsx文件
Java开发中为什么要使用spring呢?
接口测试及接口抓包常用的测试工具
基于单片机的事务管理系统
纯正体验,极致商务 | 丽亭酒店聚焦未来赛道,实现共赢发展
【正点原子】Alpha-I.MX开发板操作系统移植流程
- 原文地址:https://blog.csdn.net/qq_52813185/article/details/127588907