-
Chapter13 : Ultrahigh Throughput Protein-Ligand Docking with Deep Learning
reading notes of《Artificial Intelligence in Drug Design》
1.Introduction
- uHTVS campaigns aim well beyond compound libraries of 100 million, with the sights set on the 15.5 billion Enamine real space library and beyond.
- Compound diversity is an essential aspect of a successful uHTVS campaign.
- Humans have roughly 20 million proteins, give or take an order of magnitude for mutant and wild-type proteins, and the drug-like chemical space spans up to an estimated 1060 unique molecules.
2.Materials
2.1.Chemical Libraries
- Common chemical libraries for virtual screening are ZINC, and Enamine Real. These compound libraries are often obtained in SMILES or SDF format.
- If a set of SDF files is obtained, utilize an open source tool such as OpenBabel to covert “.sdf” to “.smi”.
2.2.Protein Preparation
- Proteins can be obtained from the Protein Data Bank (PDB). Structure preparation from this point will depend on the docking protocol used for generating scores.
2.3.Docking Protocol
- A docking protocol is a program which optimizes a scoring function by sampling positions of a 3D ligand in a protein binding region.
- An important but sometimes overlooked preparation for docking is enumerating 3D conformations of compounds in the library (see Fig. 1).

- Consider function freceptor to be a scoring function prepared with a particular protein pocket. We associate the score of the predicted pose, m a x τ ∈ v a l i d p o s e s f r e c e p t o r ( τ ) max_{\tau\in valid\ poses}f_{receptor}(\tau) maxτ∈valid posesfreceptor(τ) as a stand-in property to rank the “goodness” of molecules in a database (some authors call this value the predicted binding affinity.
- There are many scoring functions used, such as DOCK, GOLD, and FlexX.
- There is research into using the combination, or consensus score, as a more reliably and interpretable metric, such as CScore.
- A general overview of scoring functions, and outlining the differences amongst them for sampling speed is available。
3.Methods
- There is the option of ignoring the idea of scoring functions and utilizing an ML/AI model to generate the pose directly without the exhaustive search framework—such models tend to suffer from (1) their specificity to a particular scoring function protocol used for the training data, (and the imposition of a docking protocol), and (2) how docking the objective is nearly cast to mimic the physical processes involved in protein–ligand binding, which is usually represented as a multiobjective optimization problem.

3.1.Deep Learning for Protein-Ligand Docking
- With two goals in mind, accurate and informative virtual screens that extend deeply and diversely into chemical space, the tools we choose for uHTVS must be scalable for the problem.
- As Table 1 outlines, as machines become heavier on GPU compute targets, and with the rise of hardware accelerators for deep learning, adjusting the tools of uHTVS toward deep learning addresses one of the biggest problems in this space: library diversity.
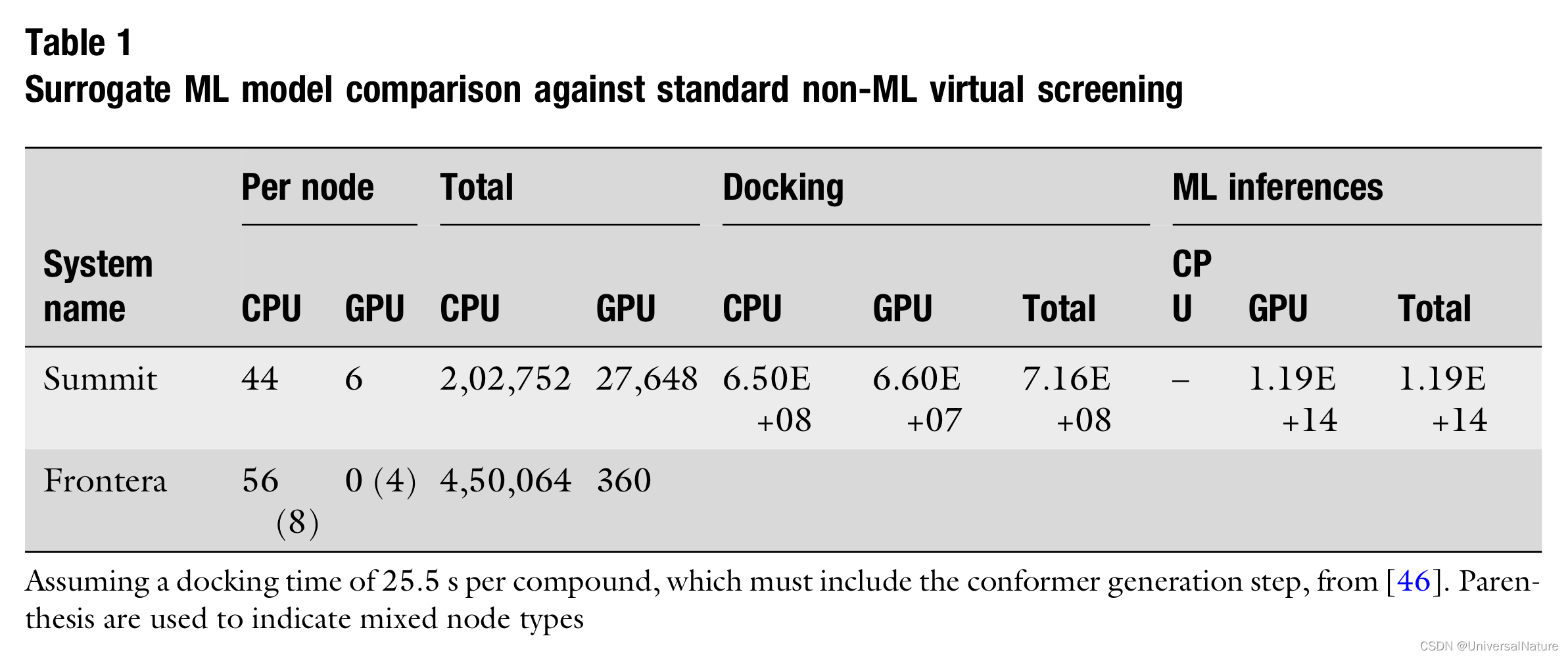
- Given this viewpoint, ML models as filtering large library to a list of hits, those molecules which pass the filter should be consid- ered for standard programmatic docking.
3.2.Model Types and Featurization
- It should be noted that molecular descriptors are distinct from of molecular fingerprints. Molecular fingerprints, another alternative for featurization, are bit vectors based on hashing different molecular neighborhoods together. Molecular fingerprints originated for use in databases as a surrogate for molecular similarity. Unlike fingerprints, molecular descriptors are explainable, where fingerprints individual bits are relatively opaque.



3.3.Analysis
- uHTVS distributions are highly skewed and vary heavily among targets and VS campaigns (see Note 2). uHTVS from docking are unlike normal distributions. In particular, one is interested in high scores rather than the mean or central tendency of data.
- If the goal is to locate 100 top scoring hits from a molecular database with one billion molecules, the model needs a predictive accuracy around 0.00001% (which should go without reference, is unheard of in machine learning literature).
- In the literature, one often encounters predictive models being evaluated based on their mean absolute error (MAE), mean squared error (MSE), or correlation coefficients (such as r2 or Pearson). In this section, we will argue these measures are uninformative at best and most often misleading and propose adopting a pragmatic model evaluation scheme based on the specifics of uHTVS.
- Scaffold splitting involves splitting molecular data into clusters based on their scaffold and holding out specific scaffolds from the training data to evaluate distribution model performance. In a similar light, time dimension splitting involves hiding compounds that were recently discovered pharmaceuticals to see if the model could have discovered them without seeing that class of molecular before.
4.Notes

-
相关阅读:
windows docker 部署 rocketmq
西门子V20变频器调试基本步骤示例
传奇出现黑屏卡屏不动是怎么回事
13年实践经验总结,200多页PPT的企业级推荐系统原理与实践,助力企业精细化与个性化运营...
springBoot异步线程,父子线程数据传递的5种姿势
【Qt】ubuntu下Qt开发环境的搭建
短视频无人直播双端开播源码部署
【苹果位置推相册推送】包括撤消受权或更改Apple软件中可访问服务的任何API)
Vue路由和Node.js环境搭建
实现PXE批量网络装机及kickstrat无人值守安装
- 原文地址:https://blog.csdn.net/weixin_52812620/article/details/126481637